Fratelli d’Italia, SVP, Freiheitliche, Lista Civica e Forza Italia votano nel Consiglio della Provincia Autonoma di Bolzano contro la necessaria revisione dell’obbligo vaccinale pediatrico prevista dalla legge e contro la necessaria trasparenza della politica vaccinale
Fratelli d’Italia in Alto Adige/Sudtirolo dimostra una volta di più la sua scandalosa ipocrisia: mentre l’Assessore Galateo vota contro legalità e trasparenza, la consigliera FdI Scarafoni – guarda caso – è rimasta assente giusto il tempo della discussione e votazione della mozione
andata oggi in discussione e votazione, e che comprendeva dei punti che ogni rappresentante del popolo che si riconosce nei principi della nostra Costituzione e rispetta e garantisce il Diritto Fondamentale dei cittadini alla necessaria trasparenza, avrebbe dovuto sostenere.
Invece, anche gli esponenti provinciali di Fratelli d’Italia, oltre ai colleghi di Forza Italia, Lista Civica, SVP e Freiheitliche, una volta di più, hanno votato contro la garanzia della legalità e trasparenza a cui i cittadini, proprio nella politica sanitaria hanno un Diritto Fondamentale.
Sin dall’introduzione dell’obbligo vaccinale pediatrico nel 2017 – con l’esclusione dalla scuola dell’infanzia, dalle strutture di assistenza alla prima infanzia e dal servizio Tagesmutter di bambini non rispondenti al piano nazionale vaccinale – il rispettivo Ministro della Salute ogni tra anni (e in caso di urgenza anche durante il periodo triennale) dopo la necessaria raccolta di dati e dell’opinione, tra le altre, anche della Conferenza Permanente per i rapporti tra lo Stato, Regioni e Province Autonome, avrebbe dovuto sottoporre ad una revisione (con esito aperto) l’obbligo vaccinale pediatrico, attualmente previsto per 10 vaccinazione (art. 1 comma 1-ter D.L. 73/2017).
Anche l’attuale governo Meloni, nella persona del Ministro alla Salute Orazio Schillaci, viola gravemente l’obbligo di rivedere (con esito aperto) l’obbligo vaccinale, omettendo, dunque, anche di raccogliere l’opinione della Conferenza Permanente per i rapporti tra lo Stato, Regioni e Province Autonome.
Il comitato di consulenza per la strategia vaccinale (NITAG) è stato sospeso sin dall’ agosto 2025, dopo che la nomina di due membri che non stanno agli ordini della lobby farmacologica, ha provocato la massiccia pressione mediatica organizzata dalla lobby dei produttori dei vaccini, e il Ministro della Salute ha preferito chiudere la questione, annullando l’operatività dell’organo, anziché garantire un organo con membri liberi da conflitti d’interessi.
Mentre in paesi come negli USA, l’organo di consulenza per la strategia vaccinale del governo (ACIP) è ora composto da esperti non finanziati dai produttori dei vaccini, e i cittadini possono seguire online le discussioni del comitato di consulenza della strategia vaccinale del governo federale e persino presentare delle domande alle cui viene data risposta dagli esperti online, in Italia il Ministro alla Salute agisce in violazione dell’obbligo di revisione e in totale difetto di trasparenza.
Mentre i cittadini degli USA apprendono nel dettaglio online le motivazioni scientifiche delle scelte di strategia vaccinale del loro governo, in Italia i genitori sono confrontati con un obbligo vaccinale che avrebbe dovuto essere rivisto già per almeno due volte e per il cui adempimento vengono usate delle sostanze, la cui efficacia e sicurezza non è mai stata confermata in studi clinici con veri gruppi di controllo, fatto peraltro ammesso dallo stesso Assessore alla Salute dell’Alto Adige e rispetto alle quali il CDC dal novembre 2025 non esclude più una possibile connessione con il numero paurosamente crescente dei casi di autismo anche in Italia!
I membri che oggi hanno votato contro la mozione, articolata in vari sottopunti, di cui i primi mai potevano essere rigettati senza esprimere di fatto l’approvazione di un operato illegittimo e gravemente intrasparente, hanno dimostrato di non voler tutelare Diritti Fondamentali dei Cittadini e hanno dimostrato di agire contro l’interesse della Provincia Autonoma di Bolzano di veder garantita la propria posizione come membro della Conferenza Permanente per i rapporti tra lo Stato, Regioni e Province Autonome.
La mozione prevedeva la votazione nominale e separata per punto sulle seguenti richieste (le complesse premesse di natura normativa, scientifica e istituzionale, le trovate nella mozione in allegato):
Il Consiglio provinciale dell’Alto Adige obblighi la Giunta provinciale
nella persona del Presidente della Provincia, a invitare immediatamente la Presidente del Governo a voler convocare, ai sensi dell’art. 12 della legge n. 400 del 23.08.1988, la Conferenza permanente per i rapporti tra Stato, Regioni e Province autonome:
1.1 ai fini della revisione dell’obbligo di vaccinazione pediatrica prevista dall’art. 1, comma 1-ter, del D.L. 73/2017
1.2 in particolare con l’obiettivo di condurre, nell’ambito della Conferenza permanente per i rapporti tra Stato, regioni e province autonome una discussione sul tema dell’obbligo vaccinale pediatrico trasparente, basata su prove scientifiche e non su propaganda dogmatica, ed accessibile ai cittadini
1.3 in particolare con l’obiettivo di chiedere nell’ambito della Conferenza permanente per i rapporti tra lo Stato, le regioni e le province autonome, un comitato consultivo libero da conflitti di interesse per le decisioni relative alla strategia nazionale di vaccinazione
1.4 in particolare con l’obiettivo di chiedere nell‘ambito della Conferenza permanente per le relazioni tra Stato, regioni e province autonome, la necessaria trasparenza, a cui i cittadini hanno un diritto fondamentale, della discussione sia sulla strategia vaccinale nazionale che sull’obbligo vaccinale pediatrico in particolare, e di richiedere, ad esempio, la trasmissione online di tale discussione
1.5 In particolare, considerando che né la sicurezza né l’efficacia dei vaccini pediatrici attualmente in uso in Italia sono state testate e confermate in studi clinici con veri gruppi di controllo, e considerando che dal 19 novembre 2025 il CDC (Centers for Disease Control and Prevention) non esclude più un nesso tra le vaccinazioni pediatriche e l’autismo, che venga sospeso immediatamente, a titolo precauzionale, l’obbligo di vaccinazione dei bambini.
1.6 in particolare con l’obiettivo di lasciare la decisione sulla vaccinazione pediatrica ai genitori, ai quali deve essere garantita un’informazione completa, obiettiva, basata su prove scientifiche e senza alcuna censura, affinché possano prendere una decisione libera e informata
fino alla decisione della Conferenza permanente per le relazioni tra Stato, regioni e province autonome, e nell’esercizio della competenza legislativa primaria in materia di scuola dell‘infanzia, di non escludere dalla scuola dell’infanzia i bambini che non sono in regola con il piano vaccinale nazionale e di provvedere all’annullamento dei decreti di decadenza dall’iscrizione alla scuola dell’infanzia già notificati alle famiglie interessate, poiché il pretesto di dover proteggere i bambini immunodepressi di fatto non è fondato, mentre è evidente la necessità di proteggere tutti i bambini dai rischi per la loro salute derivanti dalla somministrazione ripetuta di sostanze contenenti particelle di alluminio, la cui sicurezza ed efficacia non sono mai state testate in studi clinici con veri gruppi di controllo
fino alla decisione della Conferenza permanente per i rapporti tra Stato, regioni e province autonome, garantire che nessun bambino non vaccinato rimanga escluso dalle struttura di assistenza alla prima infanzia, compreso il servizio Tagesmutter, poiché il pretesto di dover proteggere i bambini immunodepressi di fatto non è fondato, mentre è evidente la necessità di proteggere tutti i bambini dai rischi per la loro salute derivanti dalla somministrazione ripetuta di sostanze contenenti particelle di alluminio, la cui sicurezza ed efficacia non sono mai state testate in studi clinici con gruppi di controllo reali.
La votazione del punto 1.1. (richiesta della revisione dell’obbligo vaccinale) non è passata per mancanza di un unico unico voto!
Il voto espresso dei singoli membri del Consiglio della Provincia Autonoma di Bolzano lo trovate in allegato.
I politici si valutano sui fatti e non sulle promesse.
In particolare i rappresentanti di Fratelli d’Italia hanno dimostrato una volta di più di fare esattamente il contrario di quanto promesso agli elettori.
La revisione dell’obbligo vaccinale pediatrico, imposta dalla legge, è attesa da tempo in Italia
La Provincia autonoma di Bolzano deve agire immediatamente e richiedere la revisione e una discussione trasparente
MOZIONE n. 351/2025 – Votazione questa settimana nel Consiglio della Provincia Autonoma di Bolzano
Ai sensi dell’art. 1, comma 1-ter, del decreto legge n. 73/2017, l’obbligo di vaccinazione dei bambini deve essere rivisto ogni tre anni, con il coinvolgimento della Conferenza permanente per i rapporti tra lo Stato, le regioni e le province autonome.
L’obbligo di vaccinazione dei bambini avrebbe già dovuto essere sottoposto ad una revisione approfondita da parte del governo italiano almeno due volte, nell’ambito di una discussione trasparente con il coinvolgimento dei rappresentanti della Provincia Autonoma di Bolzano.
Ciò non è avvenuto. Nemmeno sotto il governo Meloni.
L’attuale governo italiano si è limitato, nell’ambito della Conferenza permanente per i rapporti tra lo Stato, le regioni e le province autonome, a deliberare ossia a semplicemente prorogare di un altro anno il piano nazionale di vaccinazione PNPV 2023-2025
Vedi il punto 22 dell’accordo raggiunto il 18 dicembre 2025 nella Conferenza permanente per le relazioni tra Stato, regioni e province autonome, che prevede la proroga del piano nazionale di vaccinazione semplicemente per un altro anno.
È importante sottolineare che il piano vaccinale nazionale non prevede l’obbligo vaccinale, ma solo i vaccini raccomandati dallo Stato.
L’obbligo di vaccinazione dei bambini è stato introdotto con il decreto legge n. 73/2017 (convertito con la legge n. 119/2017) e, ai sensi dell’articolo 1, comma 1-ter, dello stesso decreto legge, deve essere sottoposto a revisione ogni tre anni o, se necessario, anche entro il periodo triennale.
Il fatto che l’obbligo vaccinale non sia previsto dal piano vaccinale nazionale, ma sia stato deciso con il decreto legge n. 73/2017, sembra non essere noto al presidente della Provincia.
Questa conclusione deriva dalla sua risposta all’interrogazione n. 39-11bis-25 presentata durante l’attuale ora delle interrogazioni.
Con tale interrogazione è stato chiesto al Presidente della Provincia se, dall’entrata in vigore il 6 agosto 2017 del decreto legge 73/2017, con il quale in Italia è stato introdotto l’obbligo vaccinale pediatrico relativo a dieci (10) vaccinazioni, sia stata invitata dal Ministero della Salute a fornire un parere ai fini di una revisione dell’obbligo vaccinale pediatrico. L’interrogazione verteva inoltre sul testo del parere della Conferenza permanente, sulla questione se l‘eventuale parere fosse stato preceduto da una discussione in seno alla Conferenza permanente e sulla posizione assunta dalla Provincia autonoma di Bolzano in merito a tale questione.
Dalla risposta scritta del presidente della Provincia
emerge chiaramente che egli fa riferimento esclusivamente al Piano Nazionale di Prevenzione Vaccinale, che però non ha nulla a che vedere con l’obbligo di vaccinazione pediatrico introdotto dal Decreto Legge n. 73/2017 e con l’obbligo previsto dall’art. 1, comma 1-ter del Decreto Legge n. 73/2017 di effettuare una revisione periodica (ogni 3 anni) e, se necessario, anche immediata, dell’obbligo di vaccinazione pediatrica.
Si deve quindi presumere che la Giunta provinciale dell’Alto Adige non sia a conoscenza dell’obbligo del Governo italiano di rivedere l’obbligo vaccinale pediatrico ai sensi dell’art. 1, comma 1-ter, del decreto legge n. 73/2017.
Il governo provinciale altoatesino deve agire immediatamente per i motivi indicati di seguito e richiedere, nell’ambito della Conferenza permanente per i rapporti tra Stato, regioni e province autonome, una revisione trasparente dell’obbligo di vaccinazione pediatrica in vigore in Italia dall’introduzione del decreto legge n. 73/2017.
L’assoluta urgenza è data, tra l’altro, dal fatto che
il piano vaccinale nazionale è stato elaborato dai politici sulla base della strategia vaccinale dettata dall’OMS,
e l’OMS è controllata dai produttori di vaccini e dai cosiddetti filantropi (Fondazione Bill & Melinda Gates), che a loro volta investono massicciamente nel business dei vaccini.
e l’obbligo di vaccinazione pediatrica vigente in Italia prevede solo pochissimi casi, determinati in ultima analisi dalla politica su suggerimento dei collaboratori dell’OMS, in cui il bambino viene esentato dall’obbligo di vaccinazione dalle autorità.
Ciò è fondamentalmente insostenibile, considerando che nessun vaccino pediatrico è stato testato in punto della sua efficacia e sicurezza in uno studio clinico con un vero gruppo di controllo.
Il fatto che non esistano studi clinici con gruppi di controllo reali (cioè gruppi di controllo a cui viene iniettato un vero placebo e nessun altro vaccino) sui vaccini pediatrici da parte delle autorità è stato confermato il 4 giugno 2024 nell’aula del Consiglio provinciale dell’Alto Adige dall’assessore alla sanità Hubert Messner in risposta a un’interrogazione, e successivamente anche dai media (vedi Neue Südtiroler Tageszeitung, L’abolizione è un must, del 10 luglio 2024).
Il dogma dell’OMS è quello di vaccinare dal grembo materno (ora è di moda vaccinare le donne incinte) fino alla bara, con un continuo ampliamento del programma vaccinale, che in 20 anni ha portato a un aumento enorme dei vaccini e delle dosi nel programma vaccinale pediatrico. E questa tendenza continua!
E sulla base di un algoritmo definito dall’OMS (WHO Causality assessment of an adverse event following immunization,
(qui il link alla versione originale in lingua inglese:
che è chiaramente a favore dei produttori di vaccini, il nesso causale con il vaccino somministrato viene escluso a priori dalle autorità sanitarie in presenza di un’altra possibile causa dell’effetto collaterale.
Nel corso degli scandalosi eventi relativi alla nomina dei membri e alla successiva sospensione del NITAG (gruppo tecnico consultivo nazionale sull’immunizzazione) italiano, è emerso che molti dei membri del NITAG italiano ricevono evidentemente denaro direttamente dall’industria farmaceutica o che gli istituti in cui operano ricevono sovvenzioni dall’industria farmaceutica
Si deve presumere che queste persone abbiano agito all’interno del NITAG come lobbisti dell’industria farmaceutica produttrice di vaccini.
Mentre negli Stati Uniti i membri del comitato consultivo per la strategia vaccinale del governo americano che ricevono contributi diretti o indiretti dall’industria farmaceutica sono stati esclusi dal comitato consultivo e sostituiti con esperti che non hanno conflitti di interesse, il Ministero della Salute italiano, dopo la nomina di due membri che criticano l‘attuale strategia vaccinale e successivamente all’enorme pressione esercitata pubblicamente dai lobbisti dell’industria farmaceutica nell’agosto 2025 contro la nomina di questi soli due membri non controllati dall’industria farmaceutica, ha semplicemente annullato il NITAG.
Attualmente in Italia non esiste un comitato consultivo ufficiale del governo sulla strategia vaccinale nazionale.
Mentre in Italia il governo, invece di garantire trasparenza e consulenti privi di conflitti di interesse per la strategia vaccinale, ha semplicemente annullato il comitato consultivo, negli Stati Uniti il comitato consultivo del governo americano per la strategia vaccinale (ACIP) è stato composto da esperti privi di conflitti di interesse e le riunioni del comitato consultivo ACIP sono trasmesse online dal CDC (Centers for Disease Control and Prevention), in modo che ogni cittadino possa farsi un’idea delle diverse opinioni e delle decisioni prese a maggioranza.
Nel corso di questa riunione di due giorni tenutasi nel dicembre 2025 dal comitato consultivo del governo statunitense sulla strategia nazionale di vaccinazione, è stata decisa la revoca della raccomandazione generale per la vaccinazione dei neonati contro l’epatite B, poiché l’epatite B è una malattia trasmissibile principalmente attraverso i rapporti sessuali o sirignhe contaminate e i bambini devono essere protetti da essa solo se la madre è risultata positiva al test dell’epatite B.
La raccomandazione generale di questa vaccinazione per tutti i neonati e i bambini piccoli è priva di qualsiasi ragionevolezza e adeguatezza, dato il profilo rischio-beneficio non positivo per la popolazione pediatrica in generale.
In Italia, tuttavia, la vaccinazione contro l’epatite B è obbligatoria, come lo era fino a poco tempo fa negli Stati Uniti.
Viene somministrato nell’ambito del vaccino esavalente (in Alto Adige è HEXYON di Sanofi-Pasteur) che viene ripetutamente iniettato ai bambini a partire dall’età di bebè.
Nel corso della riunione di due giorni dell’ACIP, trasmessa online, è emerso chiaramente che non esistono studi con veri gruppi di controllo per nessun vaccino pediatrico attualmente in uso.
A seguito di questa riunione, svolta in totale trasparenza e accessibile online ai cittadini, il presidente degli Stati Uniti ha dato mandato di rivedere con urgenza l’intero programma di vaccinazione pediatrica statunitense, dato che i risultati della strategia di vaccinazione pediatrica statunitense, che nella parte fondamentale è simile a quella italiana, sono disastrosi rispetto a quelli di paesi come la Danimarca, che prevedono molte meno vaccinazioni pediatriche e, soprattutto, nessuna vaccinazione obbligatoria.
Negli Stati Uniti, un bambino su 36 è affetto da un disturbo dello spettro autistico.
L’Italia, con un (1) bambino su 76 affetto da un disturbo dello spettro autistico (dati dell’Istituto Superiore di Sanità del 2019), non è in una situazione molto migliore
Si presume che la prevalenza dei disturbi dello spettro autistico in Italia sia ulteriormente aumentata.
Molto illuminante è la valutazione intermedia del programma di vaccinazione per bambini e adolescenti negli Stati Uniti pubblicata il 2 gennaio 2026 dalla direttrice in carica del Centro per la valutazione e la ricerca sui farmaci e membro dell’ACIP, nonché dal Chief Science and Data Officer del viceministro della Pianificazione e della Valutazione e dagli esperti del CDC, della FDA, del NIH e del CMS:
In esso gli esperti statunitensi dichiarano testualmente quanto segue:
“… come tutti i farmaci, anche i vaccini comportano dei rischi che devono essere valutati rispetto ai loro benefici. Prima e dopo l’approvazione, i produttori hanno incentivi insufficienti per studiare gli effetti collaterali dei vaccini. Le autorità di regolamentazione, come la FDA e il CDC, a volte hanno riconosciuto solo lentamente gli effetti collaterali negli studi post-marketing. La sicurezza e i rischi dei vaccini sono quindi spesso caratterizzati, quantificati o compresi in modo insufficiente. Raramente sono disponibili dati scientificamente fondati sugli effetti collaterali che consentano di determinare la relazione tra il programma di vaccinazione degli Stati Uniti e la crescente prevalenza di malattie croniche nei bambini americani. Gli interventi medici effettuati su bambini sani per prevenire le malattie e non per curarle o guarirle dovrebbero soddisfare i più elevati standard di sicurezza prima e dopo l’immissione sul mercato…
È generalmente considerato una violazione dell’etica medica di base imporre o richiedere un intervento medico, e il consenso informato è un pilastro fondamentale dell’assistenza sanitaria negli Stati Uniti e all’estero. Nel suo codice di etica medica, l’American Medical Association afferma che il trattamento medico secondo il principio del “consenso libero e informato”è fondamentale sia dal punto di vista etico che giuridico…
Un programma di vaccinazione pediatrica di successo richiede fiducia reciproca tra pazienti/genitori e medici, nonché le autorità sanitarie. Questa fiducia si basa su quattro pilastri:
Onestà scientifica in relazione ai vaccini, compreso ciò che è noto e ciò che non è noto.
Consenso informato, nessuna coercizione.
Una procedura di autorizzazione dei vaccini basata su dati scientifici comprovati e su una valutazione approfondita della sicurezza e dei rischi dei vaccini dopo l’autorizzazione.
Raccomandazioni che tengono conto delle esperienze di altri
I sistemi di monitoraggio della sicurezza dei vaccinipresentano gravi carenze, in quanto non sono sufficientemente utilizzati, tra l’altro, per valutare gli effetti a lungo termine dei vaccini, che possono essere diagnosticati e manifestarsi mesi o anni dopo la vaccinazione, nonché gli effetti di vari aspetti combinati del programma di vaccinazione.
Ad esempio, uno studio del CDC del 2023 ha rilevato che i bambini che hanno ricevuto più adiuvanti di alluminio presentavano un tasso più elevato di asma persistente, ma non sono stati condotti studi sufficienti sul legame tra vaccini e conseguenze a lungo termine sulla salute, tra cui asma, allergie, malattie autoimmuni, disturbi neurologici, otiti o altre malattie infettive.
Un programma di vaccinazione infantile efficace deve basarsi su solide conoscenze scientifiche. Ciò significa che
l’approvazione di nuovi vaccini destinati all’uso di massa dovrebbe basarsi su studi randomizzati, in doppio cieco, controllati con placebo. Finora ciò è avvenuto raramente.
Per i nuovi vaccini deve essere disponibile un sistema post-marketing per individuare rapidamente gli effetti collaterali imprevisti.
Oltre agli effetti collaterali acuti, dobbiamo valutare gli effetti a lungo termine sul sistema immunitario, come l’asma, le malattie autoimmuni, i disturbi neurologici e le infezioni aspecifiche.
Oltre ai singoli vaccini, dobbiamo valutare accuratamente la sicurezza dell’intero programma di vaccinazione, compresi gli effetti cumulativi, i tipi e gli ingredienti dei vaccini, i tempi e la sequenza delle vaccinazioni e le interazioni. L’IOM richiede da tempo studi di questo tipo.
Mentre negli Stati Uniti si sta valutando, sulla base di dati scientifici, l’efficacia dell’intero programma di vaccinazione pediatrica e sono già state apportate le prime modifiche al programma stesso, sottolineando la necessità della libera scelta in materia di vaccinazioni, il governo italiano, quantomeno dall’inizio del 2021 (scadenza del primo triennio) sta violando il suo obbligo di revisione, ovvero di verifica del programma di vaccinazione pediatrica, ed ha semplicemente alla fine del 2025 prorogato di un altro anno il programma di vaccinazione raccomandato nel 2023 dal NITAG italiano (comitato consultivo del governo per la strategia vaccinale).
E questo nonostante il NITAG sia stato annullato in agosto 2025, perché il governo italiano, a differenza di quello statunitense – preferisce evitare il confronto con il potere concentrato delle lobby dell’industria farmaceutica e continua semplicemente a perseguire la strategia vaccinale raccomandata anni fa da consulenti governativi corrotti, a porte chiuse per i cittadini, senza alcuna trasparenza e ormai anche senza consulenti pubblicamente noti per la strategia vaccinale nazionale.
Ciò è tanto più inaccettabile in quanto la più importante autorità sanitaria occidentale, il CDC (Centers for Disease Control and Prevention – la principale autorità sanitaria federale statunitense), dal 19 novembre 2025 dichiara ufficialmente sul suo sito web che non si può escludere che le vaccinazioni infantili siano una causa dell’autismo.
I rappresentanti dei partiti che compongono il governo provinciale (in primis SVP, Fratelli d’Italia) obiettano sempre che la Provincia Autonoma di Bolzano non potrebbe fare nulla contro l’obbligo di vaccinazione pediatrica. Invece, il governo provinciale non solo può agire in tal senso nell’ambito della Conferenza permanente per i rapporti tra lo Stato, regioni e province autonome, ma deve anche agire con urgenza, tenuto conto dell’obbligo previsto dal decreto legge n. 73/2017, art. 1, comma 1-ter, di rivedere l’obbligo di vaccinazione pediatrica ogni tre anni o, in circostanze eccezionali, in qualsiasi momento!
*
Alla luce di quanto sopra esposto, ho presentato la seguente mozione che andrà in votazione nel Consiglio della Provincia Autonoma di Bolzano in questi giorni:
Il Consiglio provinciale dell’Alto Adige obbllighi la Giunta provinciale
nella persona del Presidente della Provincia, a invitare immediatamente la Presidente del Governo a voler convocare, ai sensi dell’art. 12 della legge n. 400 del 23.08.1988, la Conferenza permanente per i rapporti tra Stato, Regioni e Province autonome:
1.1 ai fini della revisione dell’obbligo di vaccinazione pediatrica prevista dall’art. 1, comma 1-ter, del D.L. 73/2017
1.2 in particolare con l’obiettivo di condurre, nell’ambito della Conferenza permanente per i rapporti tra Stato, regioni e province autonome una discussione sul tema dell’obbligo vaccinale pediatrico trasparente, basata su prove scientifiche e non su propaganda dogmatica, ed accessibile ai cittadini
1.3 in particolare con l’obiettivo di chiedere nell’ambito della Conferenza permanente per i rapporti tra lo Stato, le regioni e le province autonome, un comitato consultivo libero da conflitti di interesse per le decisioni relative alla strategia nazionale di vaccinazione
1.4 in particolare con l’obiettivo di chiedere nell‘ambito della Conferenza permanente per le relazioni tra Stato, regioni e province autonome, la necessaria trasparenza, a cui i cittadini hanno un diritto fondamentale, della discussione sia sulla strategia vaccinale nazionale che sull’obbligo vaccinale pediatrico in particolare, e di richiedere, ad esempio, la trasmissione online di tale discussione
1.5 In particolare, considerando che né la sicurezza né l’efficacia dei vaccini pediatrici attualmente in uso in Italia sono state testate e confermate in studi clinici con veri gruppi di controllo, e considerando che dal 19 novembre 2025 il CDC (Centers for Disease Control and Prevention) non esclude più un nesso tra le vaccinazioni pediatriche e l’autismo, che venga sospeso immediatamente, a titolo precauzionale, l’obbligo di vaccinazione dei bambini.
1.6 in particolare con l’obiettivo di lasciare la decisione sulla vaccinazione pediatrica ai genitori, ai quali deve essere garantita un’informazione completa, obiettiva, basata su prove scientifiche e senza alcuna censura, affinché possano prendere una decisione libera e informata
fino alla decisione della Conferenza permanente per le relazioni tra Stato, regioni e province autonome, e nell’esercizio della competenza legislativa primaria in materia di scuola dell‘infanzia, di non escludere dalla scuola dell’infanzia i bambini che non sono in regola con il piano vaccinale nazionale e di provvedere all’annullamento dei decreti di decadenza dall’iscrizione alla scuola dell’infanzia già notificati alle famiglie interessate, poiché il pretesto di dover proteggere i bambini immunodepressi di fatto non è fondato, mentre è evidente la necessità di proteggere tutti i bambini dai rischi per la loro salute derivanti dalla somministrazione ripetuta di sostanze contenenti particelle di alluminio, la cui sicurezza ed efficacia non sono mai state testate in studi clinici con veri gruppi di controllo
fino alla decisione della Conferenza permanente per i rapporti tra Stato, regioni e province autonome, garantire che nessun bambino non vaccinato rimanga escluso dalle struttura di assistenza alla prima infanzia, compreso il servizio Tagesmutter, poiché il pretesto di dover proteggere i bambini immunodepressi di fatto non è fondato, mentre è evidente la necessità di proteggere tutti i bambini dai rischi per la loro salute derivanti dalla somministrazione ripetuta di sostanze contenenti particelle di alluminio, la cui sicurezza ed efficacia non sono mai state testate in studi clinici con gruppi di controllo reali.
Diritto fondamentale della popolazione al consenso libero e informato per un trattamento farmacologico violato a causa dello shedding dei cosiddetti “vaccini” contro il Covid-19
Il Governo nazionale e le Giunte delle Regioni e delle Province autonome devono agire immediatamente a tutela della popolazione!
Un nuovo studio israeliano (condotto da un team di scienziati di rinomate università israeliane) conferma la presenza dell’RNA vaccinale nella placenta, nello sperma, nel liquido seminale e nel sangue anche molti mesi dopo la “vaccinazione” con il “vaccino” Covid-19 di Pfizer (Comirnaty), ampiamente utilizzato in Israele.
Inoltre, l’RNA vaccinale di Comirnaty di Pfizer è stato trovato anche nella metà delle donne incinte non vaccinate. Ciò conferma la sua trasmissione da persone “vaccinate” a persone “non vaccinate”, il cosiddetto shedding.
Inoltre, è stata trovata l’RNA vaccinale anche molti mesi dopo l’inoculo nel corpo dei cosiddetti “vaccinati”.
Va ricordato che, secondo i documenti ufficiali di autorizzazione dei cosiddetti “vaccini” Covid-19, non sono stati effettuati studi di farmacocinetica (tra cui quelli sulla distribuzione della sostanza iniettata nell’organismo) prima dell’autorizzazione all’immissione sul mercato! Questo è semplicemente criminale!
Si veda il punto 5.2 Caratteristiche farmacocinetiche nell’allegato I alla decisione di autorizzazione all’immissione sul mercato della Commissione Europea di Comirnaty di Pfizer/BioNTech attualmente in vigore, dove per le caratteristiche farmacocinetiche è semplicemente indicato “Non pertinente”, il che significa che non sono stati condotti studi!
Così come, tra l’altro, non sono stati condotti studi sulla genotossicità e sulla cancerogenicità.
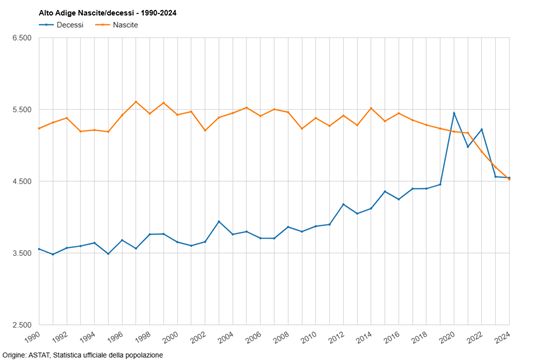
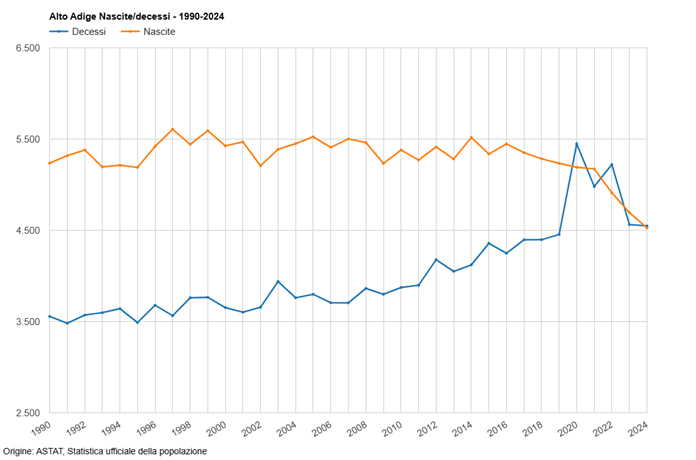
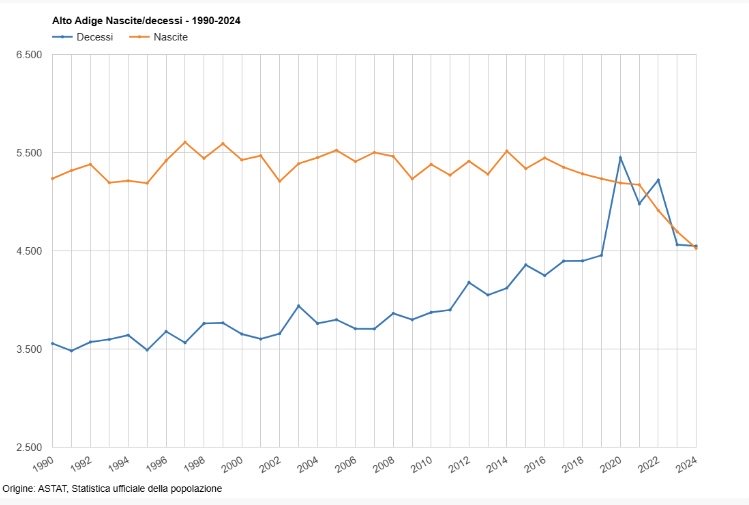
Considerato che il tasso di natalità della popolazione altoatesina/sudtirolese sta diminuendo in modo particolarmente rapido sin dall’ampia applicazione (nel 2021 e nel 2022 addirittura con “obbligo vaccinale” diretto e indiretto) di queste sostanze sperimentali basate sull’ingegneria genetica, e che anche a livello nazione la riduzione della natalità va a ritmi serrati, è inevitabile la sospensione immediata dell’uso di questi cosiddetti “vaccini” Covid-19, in particolare sulla popolazione in età fertile, inclusi i bambini!
Mentre gli Stati Uniti hanno già revocato la raccomandazione dei cosiddetti “vaccini” Covid-19, queste sostanze sperimentali altamente pericolose continuano ad essere raccomandate dal Ministero della Salute italiano e dalle autorità sanitarie nazionali e locali pure alle donne in gravidanza e in allattamento!
Già con la mia mozione n. 230/25, purtroppo respinta dalla maggioranza nel Consiglio Provinciale, avevo segnalato il drastico calo delle nascite in Alto Adige sin dall’ampia diffusione dei cosiddetti “vaccini” Covid-19 e avevo chiesto l’immediata sospensione dell’uso di queste sostanze sperimentali.
Ora è stata confermata anche la trasmissione di queste sostanze sperimentali pericolose dai “vaccinati” ai “non vaccinati”, il che non solo costituisce una violazione insostenibile del diritto fondamentale al consenso libero e informato al trattamento con un medicinale (violazione dell’art. 3 della Carta dei diritti fondamentali dell’Unione europea, art. 32 comma 2 della Costituzione della Repubblica Italiana, del Codice di Norimberga, della legge 219/2017 sul consenso libero e informato a un trattamento medico, ecc.), ma anche – considerando il fatto noto che
non sono stati effettuati studi farmacocinetici prima dell’approvazione di questi cosiddetti “vaccini” Covid-19 – una intenzionale messa in pericolo dell’intera popolazione e della sua sopravvivenza!
Pertanto, oggi ho presentato in Consiglio della Provincia Autonoma di Bolzano questa mozione:
Il Consiglio della Provincia Autonoma di Bolzano obbliga la Giunta della Provincia, considerate le più recenti accertamenti scientifici e l’evidente stato di emergenza civile dovuto allo shedding dell’RNA modificato dei cosiddetti “vaccini” Covid-19, a voler
1) nell’ambito della sua competenza primaria in materia di protezione civile, sospendere immediatamente, a titolo precauzionale, la raccomandazione della “vaccinazione” Covid-19 per l’intera popolazione in età fertile (bambini compresi)
2) provvedere alla valutazione dettagliata e trasparente e alla pubblicazione dei dati relativi agli aborti spontanei avvenuti in Alto Adige/Sudtirolo, suddivisi per trimestre o settimana di gravidanza nel rispettivo anno sin dal 2021, nonché suddivisi per donne in gravidanza trattate con un “vaccino” Covid-19 e quelle non trattate, indicando lo “stato vaccinale” Covid-19
3) far chiarire, il più possibile completo, lo “stato vaccinale” Covid-19 del partner coinvolto nel concepimento dei nati morti
4) chiedere, nella persona del governatore provinciale, immediatamente alla Presidente del governo, ai sensi dell’articolo 12 della legge n. 400 del 23 agosto 1988, l’immediata convocazione della Conferenza permanente per le relazioni tra Stato, regioni e province autonome al fine
4.1) della decisione urgente e necessaria in punto revocazione della raccomandazione della cosiddetta “vaccinazione” Covid-19 per l’intera popolazione in età fertile, in particolare per le donne in gravidanza e in allattamento nonché per i bambini
4.2) della urgente e necessaria valutazione dettagliata nonchè pubblicazione dei dati nazionali sugli aborti spontanei, suddivisi per trimestre o settimana di gravidanza nell’anno in questione dal 2021 e separatamente per soggetti trattati con un “vaccino” Covid-19 e soggetti non trattati,
4.3) nonché per il più ampio possibile chiarimento dello status vaccinale “ Covid-19” del partner nella procreazione dei nati morti
Mi auguro che si attivino subito nella stessa direzione membri del Parlamento e delle Giunte delle Regioni e della Provincia Autonoma di Trento.
RA/Avv. DDr. Renate Holzeisen
Abgeordnete zum Südtiroler Landtag – Membro del Consiglio della Provincia Autonoma di Bolzano
Inversione di rotta ufficiale del CDC: da ieri non esclude più un nesso causale tra vaccini pediatrici e autismo
Ammissione ufficiale da parte della principale autorità sanitaria degli USA di aver escluso per decenni, senza alcuna prova scientifica, un nesso causale tra vaccinazione pediatrica e autismo al solo scopo di contrastare l’esitazione vaccinale
L’obbligo di vaccinazione pediatrica in vigore in Italia definitivamente non più sostenibile!
Necessaria la immediata sospensione dell’obbligo vaccinale pediatrico e un’informazione corretta e completa dei genitori per un consenso informato e libero!
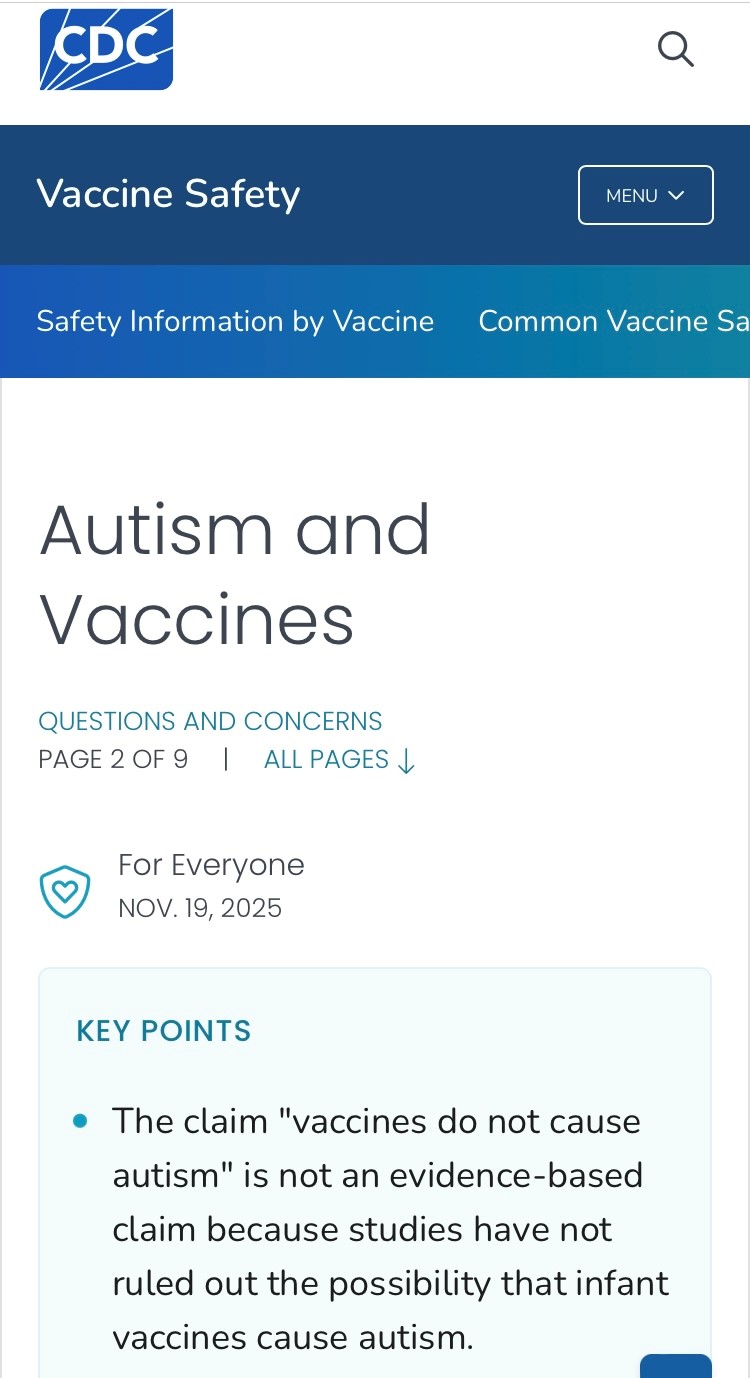
Il CDC (Centers for Disease Control and Prevention – la principale autorità sanitaria federale degli USA) da ieri dichiara ufficialmente sul suo sito web che non è possibile escludere che le vaccinazioni pediatriche siano una causa dell’autismo, compiendo così una svolta della sua posizione sul tema, attesa da tanto tempo!
L’affermazione “i vaccini non causano l’autismo” non è basata su prove scientifiche, poiché gli studi non hanno escluso la possibilità che i vaccini pediatrici causino l’autismo.
Gli studi che sostengono tale collegamento sono stati ignorati dalle autorità sanitarie.
L’HHS ha avviato una valutazione completa delle cause dell’autismo, comprese indagini sui meccanismi biologici plausibili e sui potenziali nessi causali.
Ai sensi del Data Quality Act (DQA), che impone alle agenzie federali di garantire la qualità, l’obiettività, l’utilità e l’integrità delle informazioni che diffondono al pubblico, questa pagina web è stata aggiornata perché l’affermazione “I vaccini non causano l’autismo” non è basata su prove scientifiche.Gli studi scientifici non hanno escluso la possibilità che i vaccini pediatrici contribuiscano allo sviluppo dell’autismo.
Tuttavia, questa affermazione è stata storicamente diffusa dal CDC e da altre agenzie sanitarie federali all’interno dell’HHS per prevenire l’esitazione vaccinale.
Inoltre, il CDC dichiara testualmente da ieri sul suo sito web:
“L’HHS (Department of Health and Human Services – il Ministero federale della Salute degli USA) ha avviato una valutazione completa delle cause dell’autismo, comprese indagini sui possibili meccanismi biologici e sui potenziali nessi causali. Questa pagina web sarà aggiornata con i risultati scientifici più autorevoli derivanti dalla valutazione completa delle cause dell’autismo condotta dall’HHS, come richiesto dal DQA. Di seguito, come richiesto dal DQA, vengono descritti in dettaglio lo stato delle prove e degli studi, e la loro mancanza, riguardanti i vaccini e il disturbo dello spettro autistico (autismo) e vengono delineate le future direzioni di ricerca dell’HHS per fornire delle risposte. È fondamentale affrontare le domande che gli americani si pongono sulla causa dell’autismo per garantire che le linee guida in materia di salute pubblica rispondano adeguatamente alle loro preoccupazioni. Circa uno su due dei genitori di bambini autistici intervistati ritiene che i vaccini abbiano avuto un ruolo nell’autismo dei propri figli, indicando spesso i vaccini somministrati nei primi sei mesi di vita (difterite, tetano, pertosse (DTaP), epatite B (HepB), Haemophilus influenzae tipo B (Hib), poliovirus inattivato (IPV) e pneumococco coniugato (PCV)) e uno somministrato al primo anno di vita o dopo (morbillo, parotite, rosolia (MMR)).Questa connessione non è stata studiata in modo adeguato e approfondito dalla comunità scientifica. Nel 1986, il programma di immunizzazione infantile del CDC per i neonati (≤ 1 anno di età) raccomandava cinque dosi totali di vaccini: due dosi orali di vaccino antipolio orale (OPV) e tre dosi iniettabili di vaccino contro la difterite, il tetano e la pertosse (DTP). Nel 2025, il calendario del CDC raccomandava tre dosi orali di rotavirus (RV) e tre dosi iniettabili ciascuna di HepB, DTaP, Hib, PCV e IPV entro i sei mesi di età, due dosi iniettabili di influenza (IIV) entro i 7 mesi di età e dosi iniettabili di Hib, PCV, MMR, varicella (VAR) ed epatite A (HepA) entro i 12 mesi di età. L’aumento della prevalenza dell’autismo dagli anni ’80 è correlato all’aumento del numero di vaccini somministrati ai neonati. …Ad esempio, uno studio ha rilevato che gli adiuvanti di alluminio nei vaccini avevano la più alta correlazione statistica con l’aumento della prevalenza dell’autismo tra le numerose cause ambientali sospette… il CDC ha violato il DQA quando ha affermato che “i vaccini non causano l’autismo”. Il CDC sta ora correggendo la dichiarazione e l’HHS sta fornendo finanziamenti e sostegno adeguati per gli studi relativi ai vaccini pediatrici e all’autismo.”
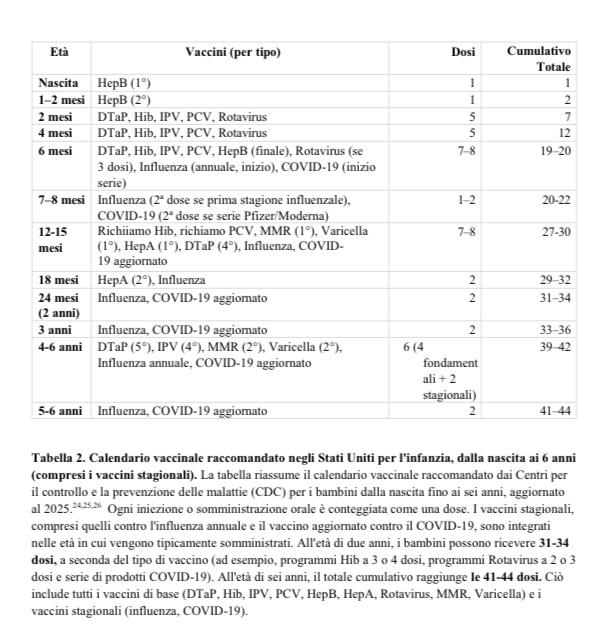
Come negli Stati Uniti, anche in Italia il tasso di autismo è esploso dall’estensione del programma di vaccinazione pediatrica e dall’introduzione dell’obbligo vaccinale pediatrico nel 2017. L’anno scorso è stato ufficialmente dichiarato che in Italia 1 bambino su 76 bambini soffre di un disturbo dello spettro autistico. E la tendenza è in continuo aumento!
Che questo aumento corrisponda ad un incremento reale dei casi è stato confermato, ad esempio, nel corso di un’audizione nell’autunno dello scorso anno dei rappresentanti delle scuole dell’infanzia e di tutti i livelli delle scuole altoatesine/sudtirolesi delle tre lingue ufficiali (tedesco, italiano e ladino) da parte della Prima Commissione Legislativa (di cui faccio parte) nel Consiglio della Provincia Autonoma di Bolzano. Alla mia domanda concreta se l’aumento del numero di bambini affetti da disturbi dello spettro autistico, lamentato dai rappresentanti delle scuole dell’infanzia e delle scuole d’obbligo, fosse dovuto a un aumento reale o a un cambiamento nel metodo di diagnosi, tutti i rappresentanti delle scuole dell’infanzia e delle scuole d’obbligo da noi invitati in Commissione Legislativa hanno risposto all’unisono che l’aumento è reale e che non si dispone di un sufficiente numero di personale di supporto!
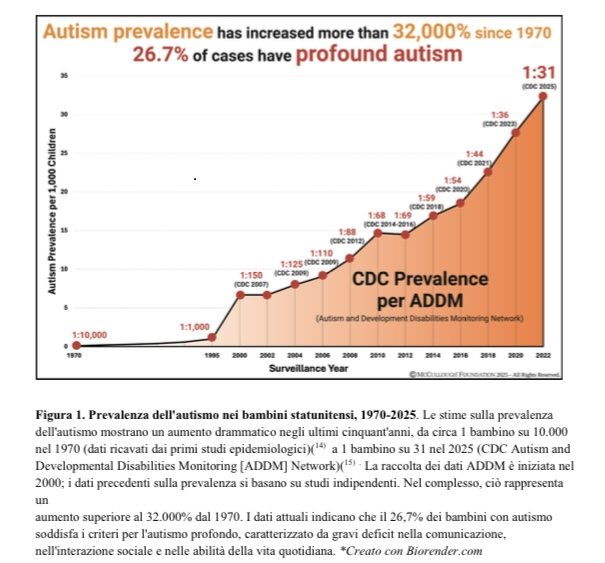
Considerando che tutti i vaccini pediatrici attualmente utilizzati in Alto Adige-Sudtirolo /Italia non sono mai stati testati in studi clinici con veri gruppi di controllo (cioè un vero gruppo placebo) per verificarne la sicurezza e l’efficacia (come ha dovuto confermare lo stesso Assessore alla Salute Hubert Messner in risposta ad una mia interrogazione in Consiglio della Provincia Autonoma di Bolzano), il mantenimento dell’obbligo di vaccinazione pediatrica (10 vaccinazioni obbligatorie in Italia) è insosteniibile alla luce delle prove scientifiche schiaccianti già disponibili (vedi la meta-analisi recentemente pubblicata dalla Peter McCullough Foundation, qui il link alla versione originale in inglese: https://mcculloughfnd.org/pages/autism-research-report e qui il link alla traduzione in lingua italiana: https://drive.google.com/file/d/12biGPpXwQTE8ZpS5XosGD9sO3DkXtnTT/viewe della conseguente modifica della posizione ufficiale dell’autorità sanitaria statunitense competente in materia, non è più sostenibile!
Chiedo al Governatore della Provincia Autonoma di Bolzano Arno Kompatscher, responsabile della protezione civile, nonché all’Assessore alla Salute Hubert Messner, di voler adottare immediatamente misure adeguate per proteggere la salute dei bambini, tra cui:
l’urgente sollecito rivolto alla Presidente del governo italiano Giorgia Meloni di voler convocare senza indugio la Conferenza permanente tra Stato, Regioni e Province autonome ai fini della revisione, prevista dall’art. 1, comma 1-ter decreto legge n. 73/2017 (convertito dalla legge n. 119/2017 – Legge Lorenzin), con l’obiettivo di abolire l’obbligo della vaccinazione pediatrica e della garanzia di un’informazione corretta ed esauriente dei genitori sull’effettivo profilo di rischio (e di efficacia) dei vaccini pediatrici attualmente in uso.
Chiedo inoltre a tutti i partiti di opposizione nel Consiglio della Provincia Autonoma di Bolzano, che finora si sono sistematicamente opposti alla messa in discussione dell’obbligo vaccinale pediatrico – in primis i Verdi, il TEAM K, ma anche il PD, la Lista Civica, Forza Italia – di volersi finalmente unire a me e agli altri troppo pochi consiglieri per difendere la salute dei bambini sulla base delle conoscenze scientifiche e della legislazione sul farmaco, perché fin adesso hanno fatto esattamente il contrario.
Chiedo in particolare anche ai consiglieri di Fratelli d’Italia, tra cui l’Assessore Marco Galateo (responsabile dell’istruzione in lingua italiana, e dunque anche delle scuole dell’infanzia) di voler dimostrare coerenza e imporre nell’ambito della coalizione di governo con la Südtiroler Volkspartei la necessaria tutela dei bambini! Ogni voto contrario alla richiesta di prendere urgenti misure contro l’obbligo vaccinale e per l’applicazione del Diritto del Farmaco (tra cui il rispetto della necessaria prescrizione medica per ogni vaccinazione e la garanzia di un’informazione corretta ed esauriente) è assolutamente scandaloso in modo particolare se proviene dai rappresentanti del partito di Giorgia Meloni, che quantomeno apparentemente sembra essere interessato ad una urgente revisione dell’obbligo vaccinale pediatrico.
La costrizione dei genitori – con l’esclusione dei loro figli non vaccinati dalle strutture di assistenza alla prima infanzia, dal servizio “Tagesmutter” e dalle scuole dell’infanzia – a somministrare ripetutamente ai propri figli (a partire dall’età di neonato) sostanze, la cui sicurezza non è stata testata e confermata, costituisce – in considerazione dell’ammissione ufficiale da parte della principale autorità sanitaria degli USA di aver escluso senza alcuna prova scientifica per decenni un nesso causale tra la vaccinazione pediatrica e l’autismo, al solo scopo di contrastare l’esitazione vaccinale – una chiara violazione del Codice di Norimberga, della Convenzione internazionale di Oviedo e degli articoli 2, 10, 13, 31, 32 e 34 (quest’ultimo articolo riguarda l’anno di scuola dell’infanzia obbligatorio in Alto Adige/Sudtirolo), nonché degli articoli della Convenzione Europea dei Diritti dell’Uomo 1(obbligo di garantire i diritti umani), 14 (divieto di discriminazione), art. 1 Protocollo n. 12 Convenzione europea dei diritti dell’uomo (divieto generale di discriminazione), degli articoli 1 (dignità umana), 3 (diritto all’integrità fisica), 14 (diritto all’istruzione), 21 (non discriminazione), 24 (diritti dei minori) della Carta dei diritti fondamentali dell’Unione Europea, nonché della Carta dei diritti dei bambini delle Nazioni Unite!
RA/Avv. DDr. Renate Holzeisen
Abgeordnete zum Südtiroler Landtag – Membro del Consiglio della Provincia Autonoma di Bolzano
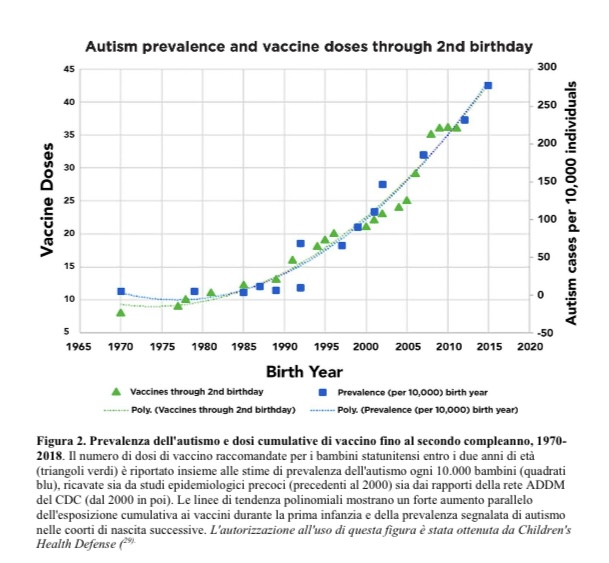
Ieri è stata pubblicata negli Stati Uniti una meta-analisi completa di tutti gli studi finora pubblicati e noti sulle cause dei disturbi dello spettro autistico.
Questo importante meta-studio, senza precedenti per le sue dimensioni, che esamina in modo approfondito gli studi epidemiologici, clinici e meccanicistici che valutano i potenziali fattori di rischio dei disturbi dello spettro autistico, giunge letteralmente alla seguente conclusione dettagliata e documentata:
“L’insieme delle prove sostiene un modello multifattoriale dei disturbi dello spettro autistico, in cui interagiscono predisposizione genetica, neuroimmunobiologia, tossine ambientali, fattori di stress perinatali ed esposizioni iatrogene.
Le vaccinazioni combinate e precoci di routine durante l’infanzia rappresentano il fattore di rischio modificabile più significativo per i disturbi dello spettro autistico, come confermato da risultati meccanicistici, clinici ed epidemiologici concordanti e caratterizzati da un uso intensificato, dall’accumulo di più dosi durante le fasi critiche dello sviluppo neurologico e dalla mancanza di ricerche sulla sicurezza cumulativa del programma vaccinale pediatrico completo.
Poiché la prevalenza dei disturbi dello spettro autistico continua ad aumentare a un ritmo senza precedenti, chiarire i rischi associati al dosaggio cumulativo dei vaccini e al momento della vaccinazione rimane una priorità urgente per la salute pubblica.
Negli Stati Uniti, 1 bambino su 36 ha una diagnosi di autismo, mentre in Italia/Alto Adige-Sudtirolo lo scorso anno la percentuale era di 1 bambino su 76! E la tendenza è in aumento!
In un’audizione tenutasi nell’autunno 2024 nel Consiglio della Provincia Autonoma di Bolzano (da parte della Prima Commissione Legislativa, di cui faccio parte) con i rappresentanti delle scuole dell’infanzia e delle scuole di ogni livello di lingua tedesca, italiana e ladina, alla mia domanda concreta se i casi di disturbo dello spettro autistico siano effettivamente aumentati o siano invece spiegabili con una diagnostica più raffinata, tutti i rappresentanti presenti hanno dichiarato all’unisono che l’aumento esplosivo rispetto al passato è effettivo e non può essere attribuito a criteri diagnostici modificati.
L’andamento della prevalenza dell’autismo in relazione alle dosi di vaccino somministrate fino al secondo compleanno, illustrato sulla base dei dati CDC nell’ampio studio META, parla chiaro!
L’Italia ha un programma di vaccinazione pediatrica sostanzialmente simile a quello degli Stati Uniti.
Quello statunitense è leggermente più ampio, da qui il tasso di autismo ancora più elevato negli Stati Uniti.
Va ricordato che nessuno dei vaccini pediatrici attualmente in uso (in Alto Adige-Sudtirolo, ai fini dell’adempimento dell’obbligo vaccinale” vengono utilizzati il vaccino esavalente HEXYON e il vaccino quadrivalente PROQUAD, oltre a una serie di vaccinazioni raccomandate) è stato testato in punto di efficacia e sicurezza nell’ambito di studi clinici con veri gruppi di controllo. L’assessore alla salute Hubert Messner lo ha confermato in risposta a una mia domanda specifica nell’aula del Consiglio provinciale e ai giornalisti.
Inoltre, tutti i vaccini pediatrici autorizzati a livello centrale dalla Commissione europea nell’Unione europea possono essere somministrati solo su prescrizione medica. Tuttavia, anche l’Azienda sanitaria dell’Alto Adige viola sistematicamente questo obbligo!
La presenza nel centro di vaccinazione di un medico, che nella maggior parte dei casi non vede nemmeno il bambino vaccinando, non corrisponde alla prescrizione medica dei vaccini, obbligatoriamente prevista dagli articoli 70 e seguenti della direttiva 2001/83/CE (recepito negli articoli 88 e seguenti del D.Lgs. 219/2006). In caso di prescrizione medica, se i genitori non sono stati informati (ad esempio sul fatto che non sono stati effettuati studi clinici con veri gruppi di controllo, che il vaccino esavalente HEXYON viene applicato illegalmente off label a bambini di età superiore ai 24 mesi), il medico si assume la responsabilità per eventuali danni conseguenti all’uso del prodotto vaccinale, come previsto dalla Legge sul farmaco.
I responsabili della sanità pubblica, in primis il governo Meloni, ma anche i responsabili locali in Alto Adige-Sudtirolo, sono chiamati, alla luce dei risultati di questo meta-studio, a sospendere immediatamente l’obbligo di vaccinazione pediatrica e a garantire un’informazione completa ai genitori ai fini di un vero consenso informato!
Il governo provinciale dell’Alto Adige-Sudtirolo deve chiedere alla Presidente del Governo Meloni, tramite il governatore, di convocare immediatamente la Conferenza permanente degli Stati, delle regioni e delle province autonome per discutere e decidere in merito alla urgente sospensione cautelativa dell’obbligo di vaccinazione dei bambini.
In caso contrario, i responsabili in politica e nella Sanità Pubblica saranno ritenuti personalmente responsabili di tutti gli ulteriori danni che ne deriveranno, poiché da questa meta-analisi risulta chiaro che è assolutamente necessario intervenire subito!
RA/Avv. DDr. Renate Holzeisen
Abgeordnete zum Südtiroler Landtag – Membro del Consiglio della Provincia Autonoma di Bolzano
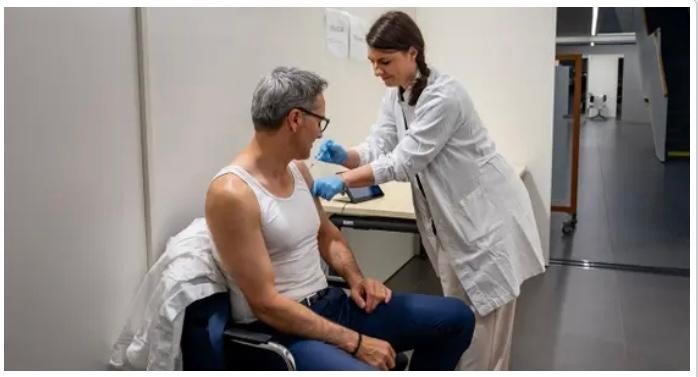
Il presidente e l’assessore alla salute della Provincia Autonoma di Bolzano in canottiera in tour promozionale per il vaccino antinfluenzale geneticamente modificato e trasmettibile nell’ambiente dalle persone vaccinate
La Commissione Europea/EMA conferma: i bambini trattati con il vaccino antinfluenzale FLUENZ di ASTRAZENECA trasmettono il virus influenzale vaccinale all’ambiente
Lo slogan pubblicitario del vaccino “proteggi te stesso e gli altri” è ridotto all’assurdo
Necessarie misure immediate di protezione civile
Il presidente della Provincia Autonoma di Bolzano Arno Kompatscher e l’assessore provinciale alla salute Hubert Messner posano in un servizio fotografico in canottiera e con la siringa posizionata, per dare il “buon esempio” alla popolazione altoatesina/sudtirolese – che per buone ragioni è molto critica nei confronti della mania dei vaccini – con l’affermazione infondata: “Il vaccino antinfluenzale è un modo semplice ed efficace per proteggere se stessi e gli altri” (così l’Assessore alla Salute Hubert Messner sul sito web dell’Azienda sanitaria dell’Alto Adige: https://www.asdaa.it/it/news/influenza-per-noi-no-proteggiti-avvio-della-campagna-vaccinale-2025), e “Vaccinarsi significa assumersi la responsabilità, sia per la propria salute che per quella della società” (così il governatore dell’Alto Adige/Sudtirolo Arno Kompatscher: https://www.asdaa.it/it/news/avanti-con-il-buon-esempio-kompatscher-e-messner-si-vaccinano).
In realtà, però, è stato dimostrato da tempo che un vaccino iniettato nella parte superiore del braccio non ha alcun effetto immunizzante sulle mucose dell’apparato respiratorio, e cioè nella portata d’ingresso del virus influenzale.
Ora l’Azienda Sanitaria dell’Alto Adige/Sudtirolo, come altre ASL sul territorio nazionale, somministra ai bambini e ai giovani di età compresa tra i 24 mesi e i 18 anni il vaccino antinfluenzale nasale FLUENZ del produttore AstraZeneca, approvato per la prima volta dalla Commissione Europea nel giugno 2024:
Questo vaccino pediatrico è prodotto mediante tecnologia genetica inversa in cellule VERO (linea cellulare ottenuta da cellule renali di cercopitechi verdi) e moltiplicato in uova di gallina fecondate. Contiene organismi geneticamente modificati (OGM). Vedi pagina 2 degli allegati alla decisione di autorizzazione del prodotto vaccinale da parte della Commissione Europea:
Al punto 4.4 (Avvertenze speciali e precauzioni d’impiegao) della versione attualmente in vigore dell’allegato I della decisione di autorizzazione della Commissione Europea, sotto il titolo “Soggetti immunocompromessi” si legge letteralmente che“Chi riceve il vaccino deve essere informato che Fluenz è un vaccino a base di virus vivi attenuati e ci può essere la possibilità di trasmissione del virus a soggetti immunocompromessi. Chi riceve il vaccino deve tentare di evitare, ogniqualvolta è possibile, il contatto ravvicinato con soggetti immunocompromessi (ad es. persone che abbiano ricevuto un trapianto di midollo osseo che devono stare in isolamento) per 1 – 2 settimane dopo la vaccinazione. Negli studi clinici la massima incidenza di recupero del virus contenuto nel vaccino si è verificata 2 – 3 giorni dopo la vaccinazione. Nei casi in cui sia inevitabile il contatto con soggetti severamente immunocompromessi, si deve valutare il possibile rischio di trasmissione del virus contenuto nel vaccino antinfluenzale rispetto al rischio di acquisire e trasmettere il virus dell’influenza da ceppi wild-type.”
Vedi pagina 4 degli allegati alla decisione di autorizzazione della Commissione Europea (link indicato sopra).
Questo è l’avvertimento ufficiale riportato direttamente nella documentazione di autorizzazione all’immissione sul mercato, che i bambini trattati con questo vaccino antinfluenzale, che contiene anche organismi geneticamente modificati, trasmettono il virus influenzale geneticamente modificato all’ambiente. e quindi infettano altre persone, anche immunocompromesse!
Lo slogan “proteggi te stesso e gli altri”, promosso dai nostri “modelli di biancheria intima” e responsabili governativi, che, invece, si comportano come “responsabili marketing dell’OMS e quindi delle grandi aziende farmaceutiche”, è così definitivamente ridotto all’assurdo!
Va ricordato che l’obbligo di vaccinazione pediatrico vigente in Italia (oggetto di procedimenti giudiziari pendenti), e che viola il Diritto del Farmaco eurounionale, viene “giustificato” con l’asserita necessità di vaccinare i bambini sani per non mettere a rischio di contagio i bambini che non possono essere vaccinati per motivi di salute.
A parte il fatto che si tratta di un’argomentazione del tutto infondata, poiché i bambini non vaccinati e immunocompromessi possono essere protetti in altri modi e in modo più efficace (come dimostrano la Svizzera e l’Austria, dove non esiste l’obbligo di vaccinazione pediatrico), l’Azienda sanitaria dell’Alto Adige sta ora coscientemente diffondendo nell’ambiente, attraverso la vaccinazione dei bambini, una carica virale pericolosa per le persone immunodepresse, come confermato dalla stessa Commissione europea/EMA, e dunque dall’Autorità Europea del Farmaco!
L’Azienda sanitaria dell’Alto Adige, insieme ai responsabili della politica sanitaria e della protezione civile della Provincia Autonoma di Bolzano (l’assessore Hubert Messner e il governatore Arno Kompatscher), sta quindi deliberatamente diffondendo un virus patogeno, disinformando gravemente la popolazione!
Si tratta di una questione grave che richiede l’immediata sospensione dell’uso di questo vaccino antinfluenzale!
Infine, va sottolineato che anche questo vaccino antinfluenzale, secondo la documentazione ufficiale di autorizzazione(vedi pagina 14 degli allegati alla decisione di autorizzazione – vedi link sopra), richiede una prescrizione medica per un uso conforme alla legge, presupposto inderogabile che l’Azienda Sanitaria dell’Alto Adige ignora costantemente e sistematicamente, violando gravemente la Legge sul Farmaco che ha efficacia di supremazia.
Tra gli effetti collaterali già riscontrati dall’autorizzazione nel giugno 2024 di questo prodotto vaccinale e menzionati nella documentazione ufficiale di autorizzazione figurano anche la sindrome di Guillain-Barré e l’encefalomiopatia mitocondriale.
È quindi ulteriormente grave che l’Azienda sanitaria dell’Alto Adige violi sistematicamente il requisito obbligatorio della prescrizione medica prevista dal Legislatore Eurounionale quale condicio sine qua non per l’uso di questo e gli altri prodotti vaccinali.
Anche per questo prodotto vaccinale nè efficacia e tantomeno la sicurezza sono state testate e confermate nell’ambito di uno studio clinico con un vero gruppo di controllo.
RA/Avv. DDr. Renate Holzeisen
Abgeordnete zum Südtiroler Landtag – Membro del Consiglio della Provincia Autonoma di Bolzano
Con grande incomprensione, io, come molti operatori sanitari (medici, ostetriche, farmacisti ecc.) ed esperti del mondo scientifico, nonché cittadini da anni molto attenti a questo tema, ho preso atto della sua affermazione secondo cui il cosiddetto “vaccino” contro il COVID-19 sarebbe sicuro soprattutto in gravidanza e durante l’allattamento e che i rischi di una “infezione da SARS-CoV-2 non vaccinata” per le donne incinte e i nascituri sarebbero di gran lunga superiori ai potenziali effetti collaterali del cosiddetto vaccino.
Per questo motivo ho inviato oggi la lettera qui allegata (tradotta dal tedesco in lingua italiana) al Prof.Dr. Christian Wiedermann, e con la quale confuto le sue affermazioni anche sulla base di nuovissime evidenze scientifiche pubblicate anche su PubMed in punto dell’evidente enorme genotossicità, cancerogenicità e mutagenicità dei cosiddetti “vaccini”-Covid-19 a mod.RNA, come la sostanza sperimentale Comirnaty di Pfizer/BioNTech, applicata attualmente in Alto Adige/Sudtirolo anche su donne incinte e allattanti.
Studi pubblicati ormai a ritmo giornaliero dimostrano che i cosiddetti “vaccini” Covid-19 a base di mod.RNA (Comirnaty di Pfizer/BioNTech, Spikevax di Moderna) contengono enormi quantità di residui di molecole di DNA e Comirnaty di Pfizer/BioNTech, inoltre, una quantità del notoriamente cancerogeno promotore-enhancer SV-40 molto superiore al limite massimo indicato da FDA e OMS.
Dallo studio pubblicato il 6 settembre 2025 su PubMed (la banca dati online del National Institute of Health del governo degli Stati Uniti) da tre eminenti scienziati statunitensi e che ha destato grande preoccupazione in tutto il mondo, perché lo studio dimostra che
i due cosiddetti “vaccini” Covid-19 a base di modRNA – Comirnaty di Pfizer/BioNTech (attualmente somministrato anche alle donne incinte e che allattano in Alto Adige/Sudtirolo) e Spikevax di Moderna – contengono da miliardi a centinaia di miliardi di molecole di DNA per dose.
Entrambe le sostanze superano di 36-627 volte le linee guida stabilite dalla FDA e dall’OMS per il DNA residuo di 10 ng/dose.
Inoltre, la sostanza Corminaty di Pfizer/BioNTech, attualmente utilizzata in Alto Adige anche su donne in gravidanza e in allattamento, supera il limite legale per il promotore-potenziatore SV40, noto per la sua elevata cancerogenicità.
Il promotore-potenziatore SV40 viene iniettato negli animali da laboratorio per produrre cellule tumorali al fine di testare farmaci antitumorali, poiché è associato a una serie di tumori maligni nell’uomo.
I cosiddetti “vaccini” a modRNA contro il Covid-19 sono quindi dimostrabilmente altamente genotossici, cancerogeni e mutageni, ovvero alterano il genoma umano, poiché l’enorme quantità di residui di DNA e il superamento del limite consentito del promotore-potenziatore SV40, notoriamente altamente cancerogeno, non significano altro.
Ed è sconcertante che il Prof. Wiedermann, in qualità di direttore dell’Istituto di medicina generale e public health della Claudiana, un centro di formazione per il personale sanitario, presenti come sicura e raccomandabile l’applicazione su donne in gravidanza e in allattamento di sostanze con un enorme potenziale di genotossicità, cancerogenicità e mutagenicità (alterazione del genoma umano).
Comirnaty di Pfizer/BioNTech, così come gli altri cosiddetti “vaccini” contro il Covid-19, non sono mai stati testati in punto genotossicità, cancerogenicità e mutagenicità, come risulta direttamente dalla documentazione di autorizzazione per l’immissione sul mercato, perché si presumeva che queste sostanze sperimentali non comportassero rischi in tal senso!
Alla popolazione sono state e continuano ad essere somministrate sostanze sperimentali altamente genotossiche, cancerogene e purtroppo anche mutagene, sulla base di una pura ipotesi di innocuità.
Vedi la pagina 30 delle attuali informazioni tecniche nell’allegato I della decisione di autorizzazione della Commissione Europea su Comirnaty di Pfizer/BioNTech:
Le donne in gravidanza erano escluse dagli estremamente scarsi ed infine anticipatamente interrotti studi clinici.
Con unostudio retrospettivo su larga scala basato sulla popolazione a Seoul, Corea del Sud, pubblicato pochi giorni fa, sono state presentate le incidenze cumulative e i rischi conseguenti di tumori in generale un anno dopo la “vaccinazione” contro il COVID-19.
I dati relativi a n. 8.407.849 persone tra il 2021 e il 2023 sono stati ricavati dalla banca dati dell’assicurazione sanitaria coreana. I partecipanti sono stati divisi in due gruppi in base al loro stato di “vaccinazione” contro il COVID-19.
I rischi di cancro alla tiroide, allo stomaco, all’intestino, ai polmoni, al seno e alla prostata sono aumentati in modo significativo un anno dopo la vaccinazione.
Ora, per la prima volta, uno studio pubblicato di recente ha dimostrato la presenza della sequenza dell’RNA “vaccinale” di Comirnaty di Pfizer/BioNTech nelle cellule tumorali di una donna di 31 anni che, dopo ripetute iniezioni di Comirnaty di Pfizer/BioNTech, ha sviluppato un tumore alla vescica ad alta malignità, cancro nel passato molto raro.
Ciò significa che l’RNA “vaccinale”, che raggiunge, impacchettato nei nanolipidi, le cellule di tutto il corpo umano, si integra nel DNA umano.
La pubblicazione di ulteriori risultati di studi sulla cancerogenicità e mutagenicità, ormai dimostrate, è stata annunciata dai ricercatori statunitensi per le prossime settimane.
Il direttore del Centro per la Salute Globale dell’Istituto Superiore di Sanità (la massima autorità scientifica sanitaria italiana), il dott. Maurizio Federico, dal 2024 mette in guardia, in articoli scientifici finanziati dal Ministero della Salute, dagli effetti collaterali causati dagli attuali “vaccini” Covid-19 e dalla loro effettiva inefficacia.
e in un collegamento diretto dall’Istituto Superiore di Sanità in una conferenza stampa tenutasi il 4 dicembre 2024 nel Consiglio della Provincia Autonoma di Bolzano, sulla base di evidenze scientifiche il Direttore del Centro Nazionale di Salute Globale presso l’ISS ha sottolineato l’inefficacia e l’alto rischio dei “vaccini” Covid-19 a base di modRNA iniettati nella parte superiore del braccio.
Il Direttore del Centro Nazionale di Salute Globale presso l’ISS ha inoltre sottolineato che è risaputo che anche il vaccino antinfluenzale non è efficace, come ha ammesso lo stesso Antony Fauci in uno studio pubblicato insieme ad altri nel 2022, perché un virus che entra attraverso le vie respiratorie non può essere bloccato da una sostanza iniettata nel muscolo del braccio!
Vedi a questo proposito la registrazione dell’interessantissimo intervento alla conferenza stampa presso il Consiglio della Provincia Autonoma di Bolzano:
Vedi qui le slide mostrate dal direttore dell’Istituto Nazionale di Salute Globale dell’Istituto Superiore di Sanità durante la conferenza stampa al Consiglio della Provincia Autonoma di Bolzano:
I timori del direttore dell’Istituto Nazionale di Sanità Globale presso l’Istituto Superiore di Sanità sono condivisi anche dal governo federale degli Stati Uniti, che già a maggio ha revocato la raccomandazione delle cosiddette “vaccinazioni” Covid-19 per le donne in gravidanza e, più recentemente, in generale.
I risultati di un nuovo studio (ancora in fase di peer-review) pubblicato da eminenti scienziati israeliani e statunitensi dei centri di ricerca statunitensi BRI e MIT, delle università di Tel Aviv e Gerusalemme (tra cui Retsef Levi, membro del nuovo comitato consultivo della FDA – Food & Drug Administration – USA) forniscono indicazioni preoccupanti su un tasso più alto del previsto di aborti indesiderati in relazione alle “vaccinazioni” mRNA-COVID-19 somministrate durante la prima fase della gravidanza (settimane 8-13).
Lo studio si riferisce a n. 226.000 gravidanze nel periodo dal 2016 al 2022. Nel periodo compreso tra il 1° marzo 2020 e il 28 febbraio 2022, n. 94.251 donne incinte hanno ricevuto una cosiddetta “vaccinazione” Covid-19 a base di mod.RNA tra l’8ª e la 13ª settimana di gravidanza. Il numero di aborti osservati era superiore del 50% rispetto agli aborti previsti e ammontava a n. 13 aborti indesiderati ogni n. 100 gravidanze. Inoltre nel 90% dei casi, le donne hanno ricevuto il cosiddetto “vaccino” Covid-19 Comirnaty di Pfizer/BioNTech (attualmente in uso anche in Alto Adige/Sudtirolo) e il resto Spikevax di Moderna.
Nello studio, gli scienziati sottolineano anche che le donne incinte erano state escluse dagli studi clinici, già di per sé scarsi e interrotti prematuramente, prima dell’approvazione dei cosiddetti studi Covid-19.
La proteina spike è stata trovata nel cordone ombelicale.
Vedi qui lo studio nella versione originale inglese:
I feti vengono quindi esposti, insieme alle donne incinte, alla tossina citotossica e cancerogena.
Anche dal piano di gestione dei rischi (RMP) del produttore, attualmente in vigore e depositato presso l’EMA, emerge chiaramente che mancano i dati di sicurezza relativi all’applicazione di Comirnaty di Pfizer/BioNTech sulle donne in gravidanza e in allattamento.
Vedi la pagina 187 dell’attuale RMP per Comirnaty di Pfizer/BioNTech:
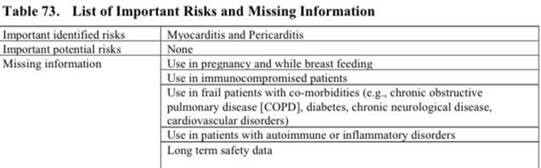
Dalle attuali informazioni al personale sanitario fornite da parte dell’EMA/Commissione Europea (e riprese dall’AIFA) su Comirnaty di Pfizer/BioNTech emerge chiaramente che la sostanza a base di mRNA modificato può causare l’infiammazione del muscolo cardiaco anche con esito fatale.
In occasione della risposta data il 9 settembre 2025 nell’aula del Consiglio della Provincia Autonoma di Bolzano alla mia domanda posta sulle donne incinte “vaccinate” contro il Covid-19, l’Assessore alla salute dell’Alto Adige/Sudtirolo (Hubert Messner) ha fatto riferimento al caso di una donna incinta che, dopo la “vaccinazione” contro il Covid-19 con una sostanza a base di RNA modificato, ha contratto la miocardite ed è stata ricoverata in ospedale.
Dato che il prof. Wiedermann è medico, dovrebbe sapere che le cicatrici del muscolo cardiaco causate dall’infiammazione non guariscono mai completamente e che molti cardiologi sottolineano la conseguente riduzione dell’aspettativa di vita.
Vedi pagina 5 dell’attuale versione dell’allegato I alla decisione di esecuzione della Commissione europea relativa all’autorizzazione all’immissione in commercio di Comirnaty di Pfizer/BioNTech:
Proprio come le donne incinte possono sviluppare la miocardite e altre malattie gravi (ad esempio il cancro – vedi sotto) a causa del “vaccino” Covid-19, il feto nel grembo materno è esposto direttamente alla proteina spike tossica, come conferma uno studio peer-reviewed: i nanolipidi iniettati con i cosiddetti “vaccini” Covid-19 a base di modRNA, in cui è racchiuso l’RNA modificato, superano facilmente la barriera placentare e, negli esperimenti sugli animali, raggiungono il feto entro un’ora, si accumulano nei suoi organi e sviluppano in modo incontrollato in tutte le cellule del feto la proteina spike tossica che porta alla morte cellulare. I timori peggiori sono stati quindi confermati da tempo.
Ecco la versione originale dello studio in lingua inglese:
Questo spiega il brusco calo delle nascite registrato dal 2021 nei paesi con un alto “tasso di copertura vaccinale” contro il Covid-19.
Anche in Alto Adige/Sudtirolo, come è noto, si registra un calo particolarmente forte delle nascite dal 2022. In Alto Adige/Sudtirolo, la vaccinazione di massa della generazione in età fertile è iniziata solo nell’estate del 2021.
Il brusco calo non può essere spiegato solo con problemi economici e sociologici (che esistevano già negli anni precedenti).
Purtroppo non sono disponibili analisi dettagliate sugli aborti spontanei in Alto Adige/Sudtirolo
che hanno dimostrato di non proteggere da un’infezione virale,
che non generano alcuna risposta immunitaria efficace nei polmoni,
che contengono enormi residui di plasmidi di DNA e un contenuto di SV-40-promotore-enhancer che supera di gran lunga il limite consentito,
che presentano effetti collaterali gravi già noti (infiammazione del muscolo cardiaco, anche con esito fatale) e molti effetti collaterali ancora poco studiati, e di cui non si conoscono gli effetti a lungo termine (come dichiarato anche dai produttori nei loro RMP)
e che, superando la barriera placentare e attraverso il cordone ombelicale, vengono trasmesse al feto, come è stato dimostrato,
significa esporre in modo del tutto irresponsabile il feto, nel quale avviene un processo di divisione cellulare particolarmente rapido e delicato, a un enorme rischio genotossico, cancerogeno e mutageno.
A causa degli effetti evidentemente catastrofici di queste sostanze sperimentali sul nostro tasso di natalità, sullo sviluppo del cancro e quindi sullo sviluppo della popolazione e sulla sua salute, la dichiarazione ufficiale della presunta innocuità da parte del Prof.Dr. Christian Wiedermann è semplicemente inconcepibile e difficilmente superabile in termini di irresponsabilità!
Infine, va ricordato che
Ai fini della legittima applicazione di ogni vaccino è richiesta la prescrizione medica inderogabilmente imposta dal Legislatore del farmaco e nelle decisioni di autorizzazione del farmaco.
Vedi ad esempio l’allegato II, punto B), della decisione di autorizzazione della Commissione europea per il cosiddetto “vaccino” Covid-19 Comirnaty di Pfizer/BioNTech
Lo stesso è riportato nella Determina dell’AIFA, con la quale viene recepita in Italia l’autorizzazione all’immissione in commercio concessa in via centralizzata dalla Commissione europea con effetto per l’intera UE.
L’obbligo di prescrizione medica per una sostanza da applicare in via parenterale è previsto dall’art. 71 della direttiva UE 2001/83/CE e dall’art. 88 del D.Lgs. 219/2006.
In Italia solo il medico è autorizzato a prescrivere un medicinale.
Il piano vaccinale nazionale e le raccomandazioni delle autorità sanitarie nazionali e locali in materia di vaccinazioni non sostituiscono la prescrizione medica (che deve essere effettuata ai sensi degli articoli 4 e 13 del codice di deontologia medica), poiché non si riferiscono né ai prodotti vaccinali specifici da utilizzare, né al soggetto specifico da vaccinare, ma a una popolazione anonima.
Il medico non deve essere soggetto ad alcun interesse, imposizione o condizionamento nell’esercizio della sua attività.
Per prescrivere un medicinale (nel caso specifico un cosiddetto vaccino), il medico deve conoscere il profilo di efficacia e di rischio del medicinale e informare il paziente, e non agire come un agente di marketing per l’industria farmaceutica.
Anche attualmente si “vaccina” sistematicamente in grave violazione della necessità di una prescrizione medica stabilita dal legislatore del farmaco per buoni motivi!
Questa grave violazione della Legge sul farmaco, in combinazione con la sistematica disinformazione e l’inganno della popolazione, ovvero l’incredibile e sfacciata negazione di rischi evidentemente enormi (fino alla morte), significa responsabilità di natura civile e penale che diventerà effettiva quando anche la giustizia, a causa della mole e della gravità delle prove che si accumulano costantemente, non potrà più sottrarsi alla sua funzione e responsabilità.
Ulteriori informazioni dettagliate le trovate nella lettera aperta qui allegata.
Negli Stati Uniti il presidente Trump e i responsabili della sanità pubblica del governo federale americano lanciano l’allarme a causa delle evidenze sempre più consistenti del nesso causale tra i vaccini pediatrici e l’epidemia di autismo
Anche in Alto Adige-Sudtirolo/Italia il tasso di autismo sta aumentando rapidamente, ma qui si continua a ignorare l’elefante nella stanza
In unaconferenza stampa tenuta nella notte (CET) tra ieri e l’altro ieri dal presidente Trump e da tutti i responsabili della sanità pubblica del governo federale statunitense, è stato sollevato l’enorme problema dell’aumento esplosivo dei casi di autismo.
Negli Stati Uniti, in media 1 bambino su 30 è affetto da un disturbo dello spettro autistico, in alcune zone come la California addirittura 1 maschio su 12.
In Italia e in Alto Adige/Sudtirolo, secondo i dati resi pubblici lo scorso anno, il tasso di autismo è di 1 bambino su 76. Anche questo è un dato estremamente preoccupante e la tendenza mostra un continuo peggioramento.
Va ricordato che in un’audizione tenutasi nell’autunno 2024 nel Consiglio della Provincia Autonoma di Bolzano, i rappresentanti delle scuole dell’infanzia e delle scuole di ogni livello dell’Alto Adige/Sudtirolo, su mia specifica richiesta, hanno confermato che l’aumento del tasso di autismo è reale e non è dovuto a criteri diagnostici modificati.
In effetti, come mi viene costantemente confermato dal personale scolastico, non si riesce più a stare al passo con la necessità di personale di supporto!
Il presidente Trump, in considerazione dell’enorme problema che riguarda non solo le famiglie colpite, ma anche il futuro della società, ha parlato chiaro nella conferenza stampa.
Egli constata che questo enorme aumento dei casi di bambini con disturbi dello spettro autistico dall’introduzione del programma di vaccinazione pediatrica in continua espansione (con obbligo di vaccinazione anche negli Stati Uniti) può essere spiegato solo con sostanze introdotte dall’esterno nel corpo dei bambini.
Trump ha dichiarato letteralmente che è inconcepibile che ai neonati vengano somministrati vaccini multivalenti (cioè vaccini combinati) pochi giorni o settimane dopo la nascita.
Ha detto testualmente: “Si pompano in questi piccoli bambini una miriade di sostanze, come se fossero cavalli. Il buon senso ci dice che questo non è giusto”.
Trump ha anche spiegato che in America ci sono gruppi di popolazione, come gli Amish, che non fanno vaccinare i propri figli e sono generalmente molto cauti nell’uso dei farmaci, e che i loro figli non sono affetti da autismo. Il ministro della Salute statunitense Robert Kennedy Jr. ha confermato che gli studi lo dimostrano.
Il presidente degli Stati Uniti ha infine dichiarato che
non vogliono vaccini pediatrici che contengano alluminio o altri metalli come adiuvanti. Il fatto è che attualmente i vaccini pediatrici contengono alluminio e altri metalli e l’alluminio ecc. – poiché iniettato e non assorbito attraverso il tratto gastrointestinale – supera la barriera emato-encefalica e ha un effetto tossico comprovato.A proposito, non esiste alcuno studio clinico sui vaccini pediatrici con un vero gruppo di controllo che riceve una sostanza senza alluminio o altri adiuvanti metallici, che abbia dimostrato la sicurezza dei vaccini pediatrici!
Le vaccinazioni, se proprio devono essere somministrate, dovrebbero essere monovalenti, al fine di ridurre il più possibile gli effetti collaterali. Negli Stati Uniti, l’ACIP (comitato consultivo di esperti per la strategia vaccinale del governo) ha già suggerito alcuni giorni fa di utilizzare il vaccino contro la varicella solo come vaccino monovalente, poiché può avere effetti collaterali gravi anche da solo.
Attualmente in Italia ai bambini viene somministrato un vaccino esavalente (difterite, tetano, pertosse, epatite B, poliomielite e Haemophilus influenzae tipo b) e un vaccino quadrivalente (morbillo, parotite, rosolia, varicella) per la somministrazione di 10 diverse vaccinazioni obbligatorie. A questi si aggiungono poi una serie di vaccinazioni raccomandate, che nella maggior parte dei casi vengono somministrate contemporaneamente! È una vera follia! Ai bambini viene somministrato un cocktail di anticorpi e additivi metallici come l’alluminio che sovraccarica il sistema immunitario di molti di loro e può causare danni neurologici permanenti.
Molti bambini reagiscono con febbre e dolori, che vengono poi “curati” dal nostro sistema sanitario con la somministrazione, in alcuni casi fatali, di paracetamolo (da noi Tachipirina).
Poiché l’epatite B può essere trasmessa solo attraverso il contatto sessuale, è assurdo vaccinare tutti i bambini contro questa malattia. In Italia/Alto Adige, invece, i bambini vengono vaccinati con il vaccino esavalente (come ad esempio Hexyon).
Il presidente degli Stati Uniti e i responsabili del settore sanitario statunitense hanno dichiarato che sono in corso studi approfonditi, ma che molti elementi indicano già un collegamento con i vaccini pediatrici e che, secondo il presidente Trump, non era più possibile attendere oltre per avvertire la popolazione.
Inoltre, è stato emesso un avviso di sicurezza in merito all’uso del paracetamolo (negli Stati Uniti Tylenol, in Italia Tachipirina) durante la gravidanza e nei bambini piccoli.
Va ricordato che da noi, proprio in caso di febbre e dolori dopo la vaccinazione, ai bambini viene somministrata la Tachipirina, ovvero il paracetamolo, su raccomandazione dei medici! Secondo le autorità sanitarie statunitensi, ciò può portare a conseguenze fatali, tra cui l’autismo!
Dopo decenni di lotte da parte dei genitori di bambini autistici, che riferiscono che i loro figli hanno sviluppato l’autismo dopo la vaccinazione, e dopo alcune sentenze giudiziarie che hanno già stabilito questa correlazione, ora il governo statunitense, per la prima volta in modo ufficiale e per mezzo del presidente degli Stati Uniti, ha dichiarato che i vaccini pediatrici sono sulla watch list per essere un fattore di rischio per l’esplosione dei casi di autismo e ha affermato che è necessario intervenire con urgenza.
Di fatto, il presidente degli Stati Uniti Trump ha lanciato un chiaro avvertimento durante la conferenza stampa. Guarda qui la registrazione:
Nel frattempo, in Alto Adige/Sudtirolo e in Italia in generale si continua a non voler vedere il cosiddetto elefante nella stanza.
In questo modo, i politici e i responsabili del settore sanitario si assumono personalmente la responsabilità del fatto che sempre più bambini soffrano di disturbi dello spettro autistico, malattie autoimmuni, ecc.
Non è un caso che ieri il presidente degli Stati Uniti sia apparso davanti ai media con questo di fatto avvertimento di protezione civile.
Il 9 settembre, infatti, durante un’audizione al Senato degli Stati Uniti, è stato presentato un importantestudiocondotto dagliesperti dell’Henry Ford Health System di Detroit (noti sostenitori dei vaccini) su un campione di bambini non vaccinati rispetto a quelli vaccinati, condotto nel periodo dal 2000 al 2016.
Da questo studio reso pubblico al Senato degli Stati Uniti emerge che, in media, i bambini vaccinati hanno una probabilità due volte e mezzo maggiore di sviluppare una malattia cronica.
Vedi in allegato lo studio tradotto in lingua italiana.
Gli autori dello studio sono noti sostenitori delle vaccinazioni e hanno dichiarato che non avrebbero osato pubblicare lo studio perché temevano per il proprio posto di lavoro e altre ritorsioni. Questo è lo stato della scienza nel XXI secolo. Ora, però, lo studio è stato reso pubblico ufficialmente in un’audizione del Senato degli Stati Uniti.
Per nessun vaccino pediatrico attualmente in uso è stata dimostrata l’efficacia e la sicurezza in studi clinici con un vero gruppo di controllo, come ha confermato l’Assessore alla Salute della Provincia Autonoma di Bolzano Hubert Messner in risposta alla mia specifica richiesta in Consiglio provinciale e in un’intervista al quotidiano Südtiroler Tageszeitung lo scorso anno (vedi Südtiroler Tageszeitung del 10.07.24 “Abschaffung ist ein muss”).
Va inoltre ricordato che ai bambini vengono somministrati i vaccini senza la prescrizione medica, invece necessaria inderogabilmente per ogni prodotto vaccinale attualmente in uso come previsto nella rispettiva decisione di autorizzazione e dalla Legge sul Farmaco!
La politica a livello locale in Alto Adige-Sudtirolo e a livello nazionale è chiamata a “tirare il freno a mano” immediatamente, come sta facendo il governo statunitense.
Come prima cosa, l’obbligo vaccinale pediatrico deve essere sospeso immediatamente.
Ciò, peraltro, sarebbe una conseguenza automatica, se la legislazione farmaceutica europea e recepita dall’Italia fosse applicata correttamente.
Infatti, la necessità di una prescrizione medica, come richiesto dalle decisioni di autorizzazione all’immissione in commercio della Commissione Europea sulla base della legislazione farmaceutica dell’UE per l’uso dei vaccini pediatrici, comporterebbe automaticamente questa conseguenza.
Inoltre, i genitori devono essere informati in modo esauriente sull’efficacia e la sicurezza dei prodotti vaccinali applicati, cosa che attualmente non avviene. Al contrario, i genitori sono costretti dall’obbligo vaccinale pediatrico (con l’esclusione dei bambini non vaccinati dalle strutture per la prima infanzia, dal servizio “Tagesmutter” e dalla scuola dell’infanzia) a far somministrare ai propri figli sostanze di cui non conoscono assolutamente il profilo di rischio. Una situazione del tutto insostenibile!
Nel frattempo, diversi Stati degli Stati Uniti, come la Florida, hanno già annunciato settimane fa l’abolizione di tutti gli obblighi vaccinali.
Ordine dei Medici italiani istruiscono i medici nel come agire per il marketing dei prodotti vaccinali, anziché informare i pazienti su efficacia e profilo di sicurezza dei prodotti vaccinali
Diffamazione da parte degli Ordini dei Medici (nello specifico di Bolzano) del Segretario alla Salute degli USA e del suo team di scienziati di massimo livello, mentre gli Ordini dei Medici negano persino il contenuto dei foglietti illustrativi dei prodotti vaccinali
Situazione inaudita che va chiarita urgentemente a livello istituzionale!
Increduli per quanto li venne inoltrato dalla Presidenza dell’Ordine dei Medici di Bolzano, tanti medici mi hanno inviato il testo di invito ad un evento sulle vaccinazioni che ebbe luogo ieri sera presso la sede dell’Ordine di Bolzano.
Tra le altre, la Presidente dell’Ordine dei Medici di Bolzano (Astrid Marsoner) nell’invito scrive questo:
“Non è solo con l’epidemia di coronavirus che abbiamo assistito a un’apparente separazione tra diritti e doveri nello Stato previdenziale e assistenziale moderno. C’è pure chi, di fronte all’appello a solidarizzare con i più vulnerabili e deboli della società, ha intravisto una forma di “fascismo” statale, un’accusa sarcastica alla luce di quanto storicamente accaduto.”
Con ciò la Presidenza dell’Ordine dei Medici di Bolzano continua a negare il fatto che i cosiddetti “vaccini”-Covid-19 mai erano stati autorizzati per prevenire la trasmissione virale e che di fatto non avevano tale efficacia! Ma ciò agli Ordini dei Medici – quanto meno qua a Bolzano – non interessa!
“Negli Stati Uniti abbiamo oggi con Robert F. Kennedy junior come Ministro della Salute, un politico, che diffonde teorie cospiratorie e complottiste sui vaccini scientificamente confutati e vuole tagliare massicciamente i fondi destinati ai relativi programmi scientifici.”
Gli Ordini dei Medici italiani – che negano persino il contenuto dei foglietti illustrativi dei prodotti vaccinali e che violano sistematicamente l’obbligo imposto dalla legge sul farmaco eurounionale e nazionale della prescrizione dei vaccini ai fini della loro legittima applicazione, arrivano a diffamare il Segretario alla Salute degli USA (e il suo team di scienziati con curricula al massimo livello mondiale) di essere complottista e negazionista… siamo arrivati a questo misero livello di strumentalizzazione degli Ordini dei Medici in Italia da parte dell’OMS e una politica che continua a navigare nelle vecchie torbidissime acque!
“In Italia, la nomina di due membri del NITAG (=gruppo di esperti statali che fornisce assistenza tecnica al Ministero della Salute), noti per essere contrari alle vaccinazioni, è stata revocata solo dopo le forti critiche da parte degli esperti del settore.”
Agli Ordini dei Medici italiani non interessa il dibattito scientifico – anzi, collaborano come longa manu dell’OMS nel totalitarismo che silenzia voci dissenzienti di esperti! Va ricordato che attualmente le nomine al NITAG sono sospese a tempo inderterminato, dopo che è stato rivelato che una buona parte dei membri graditi al sistema vaccinale sono a busta paga di big pharma.
“La salute va difesa con rigore scientifico” ha affermato Filippo Anelli, presidente della FNOMCEO, “essere medici significa anche combattere le fake news e, sopratutto, attenersi nella pratica professionale alle evidenze scientifiche.”
È proprio Filippo Anelli, il presidente della FNOMCEO, il primo a disconoscere sistematicamente le evidenze scientifiche e a precludersi ad ogni serio dibattito scientifico.
Probabilmente sarà stato proprio lui a chiedere a tutti gli Ordini dei Medici di divulgare inviti di stampo totalitario di questo genere.
“Durante questa serata discuteremo su come trasmettere queste conoscenze anche alla popolazione, ai nostri pazienti, e contrastare così il crescente scetticismo nei confronti delle vaccinazioni.”
Gli Ordini dei Medici, come in questo caso quello di Bolzano, operano evidentemente da istruttori dei medici nel come operare ai fini del marketing di big pharma, anziché informare i pazienti sull’efficacia e il profilo di sicurezza/rischio dello specifico prodotto vaccinale oppure che viene definito tale!
Ed è proprio ciò che l’OMS, controllata da GAVI e dunque dai produttori dei vaccini, vuole … e in Alto Adige/Sudtirolo e anche in altre regioni, l’Ordine dei Medici esegue questo diktat a danno dei pazienti! Che scandalo, che situazione insostenibile che deve essere accertata e fermata con la massima urgenza istituzionalmente sia a livello locale, sia a livello nazionale!
Tra il piccolo gruppo di medici che hanno presenziato all’evento sulle vaccinazioni che ebbe luogo ieri presso la sede di Bolzano (evidentemente non ha fatto tanto presa il “richiamo all’ordine da parte della Presidenza dell’Ordine in tema vaccinale ….), c’erano anche rappresentanti dell’associazione “Medici ippocratici Ärzte” (MIA), con il loro presidente Dr.med. Rudolf Schöpf, che hanno cercato di instaurare un dibattito scientifico sul tema vaccini, consegnando ai loro colleghi l’informazione succinta che oggi è stata trasmessa in via elettronica alla Presidenza dell’Ordine dei Medici di Bolzano con la richiesta di voler inoltrare l’informazione e documentazione a tutti i colleghi iscritti all’Ordine dei Medici di Bolzano, affinchè si possa instaurare un serio dibattito su questo tema importante delle vaccinazioni, visto innanzitutto l’obbligo vaccinale pediatrico.
Rimane da vedere se la Presidenza dell’Ordine dei Medici di Bolzano vorrà corrispondere a questa più che legittima, anzi, necessaria richiesta.
Il 9 settembre 2025, nell’ambito di un’audizione al Senato degli Stati Uniti è stato reso pubblico e presentato uno studio condotto dai responsabili dell’Henry Ford Health System (Detroit – USA) – sostenitori assoluti delle vaccinazioni – nel periodo dal 2000 al 2016 su un gruppo di 18.468 bambini e con cui hanno confrontato i risultati di salute a breve e lungo termine dei bambini sottoposti a una o più vaccinazioni con quelli dei bambini non sottoposti a vaccinazioni pediatriche.
Lo studio ha rilevato che, rispetto ai bambini non sottoposti a vaccinazione, la vaccinazione pediatrica era associata a un aumento complessivo di 2,5 volte della probabilità di sviluppare una malattia cronica. Questa correlazione era principalmente attribuibile ad asma, malattie atopiche, eczema, malattie autoimmuni e disturbi dello sviluppo neurologico. Dunque, la vaccinazione pediatrica può aumentare la probabilità di sviluppare una malattia cronica.
Per i dettagli, consultare lo studio allegato, disponibile anche nella traduzione in lingua italiana.
Lo studio non era stato pubblicato dagli autori della ricerca, perché temevano di perdere il posto di lavoro. Questo la dice lunga sul dibattito “scientifico” nel XXI secolo!
delle più recenti evidenze scientifiche peer-reviewed in punto quantità dei residui di plasmide DNA estremamente elevati (fino a 700 volte i limiti indicati dalla FDA) nei cosiddetti “vaccini” Covid-19 a base di modRNA (Comirnaty di Pfizer/BioNTech e Spikevax di Moderna) e della quantità del promotore SV-40, indiscutibilmente cancerogeno, che supera di gran lunga il limite massimo fissato dalle autorità regolatrici (riguarda la sostanza Comirnaty di Pfizer/BioNTech attualmente utilizzata anche in Alto Adige/Sudtirolo), motivo di grande preoccupazione a livello internazionale (vedi dettagli e allegati nella mozione, questo vale anche per i punti seguenti)
2. della conferma peer-reviewed già da quasi un anno, che questi cosiddetti “vaccini” contro il Covid-19, dopo essere stati iniettati in donne incinte, raggiungono il feto entro un’ora dall’inoculazione
3. del fatto – confermato dai dati attuali dell’ASTAT – che il tasso di natalità in Alto Adige è in rapido calo dal 2022
dei risultati della valutazione di dati dettagliati e significativi del CDC sul tasso di natalità, sulla mortalità neonatale e infantile (che dopo decenni di calo è in aumento dall’avvio della “campagna vaccinale” Covid-19), che fanno temere un impatto negativo intergenerazionale della cosiddetta “vaccinazione” contro il Covid-19
del fatto che i produttori di questi cosiddetti “vaccini” Covid-19 a base di modRNA continuano a dichiarare esplicitamente nei loro piani di gestione dei rischi che non dispongono di dati sulla sicurezza relativi alle donne in gravidanza e in allattamento
del fatto che negli Stati Uniti, nel maggio 2025, la raccomandazione della “vaccinazione” Covid-19 per le donne in gravidanza e in allattamento è stata ritirata per i motivi sopra citati e che, nel complesso, gli investimenti nella tecnologia modRNA per le malattie infettive virali (Covid-19, influenza) da parte del governo degli USA sono stati sospesi, poiché il rischio per la salute e la vita associato a queste sostanze sperimentali supera di gran lunga la presunta efficacia
del fatto che l’Alto Adige/Sudtirolo avrebbe margine di manovra sia dal punto di vista dell’autonomia di cui gode (competenza primaria nella protezione civile), sia nell’ambito della Conferenza permanente tra Stato, regioni e province, per proteggere attivamente la popolazione da conseguenze negative che si ripercuotono su più generazioni
affinchè, il Consiglio della Provincia Autonoma di Bolzano obblighi la Giunta provinciale, sulla base delle più recenti conoscenze scientifiche e dell’evidente stato di emergenza civile
1) nell’ambito della competenza primaria in materia di protezione civile, a sospendere immediatamente in via precauzionale la raccomandazione della “vaccinazione” Covid-19 per le donne in gravidanza, in periodo post-partem e in allattamento
2) nell’ambito della sua competenza primaria in materia di protezione civile, a sospendere immediatamente in via precauzionale la “vaccinazione” Covid-19 della popolazione in età fertile
3) in persona del Presidente della Provincia, a chiedere subito alla Presidente del Governo ai sensi dell’art. 12 della legge n. 400 del 23.08.1988 di voler convocare con urgenza la Conferenza permanente per le relazioni tra lo Stato, le regioni e le province autonome al fine di
3.1. deliberare con urgenza la necessaria revoca della raccomandazione della cosiddetta “vaccinazione” Covid-19 per le donne in gravidanza, in periodo post-partem e in allattamento
3.2. decidere urgentemente la sospensione generale della “vaccinazione” Covid-19 per la popolazione in età fertile
3.3. provvedere all’urgente e necessaria valutazione dettagliata e alla pubblicazione dei dati relativi agli aborti spontanei – suddivisi per trimestre di gravidanza o settimana nel rispettivo anno dal 2021 e separatamente per donne trattate con il “vaccino” Covid-19 e quelle non trattate.
L’Assessore altoatesino/sudtirolese alla salute, il medico Hubert Messner, ieri in Aula ha persino esplicitamente negato il rapido calo del tasso di natalità in Alto Adige/Sudtirolo dal 2022, nonostante questo risulti chiaramente dai dati ASTAT (Istituto Statistico della Provincia Autonoma di Bolzano
Mi posso spiegare questa reazione alla mia mozione solo con una dissonanza cognitiva o la mancanza di buona fede.
E il consigliere del TEAM-K e medico Franz Ploner ha affermato esplicitamente nella sua dichiarazione contraria alla mia mozione che sarebbe dimostrato che i cosiddetti “vaccini” Covid-19 sono sicuri per i nascituri!
Il governatore dell’Alto Adige/Sudtirolo Kompatscher – una volta di più – non intende esercitare i poteri che ci spettano in virtù della nostra autonomia e si rifiuta persino di sottoporre questo importante tema alla Conferenza permanente tra Stato, Regioni e Province autonome, cosa che non solo potrebbe fare in qualsiasi momento, ma che – alla luce dei dati allarmanti – deve fare!
La maggioranza del Consiglio della Provincia Autonoma di Bolzano, ovvero l’intero governo provinciale e i relativi consiglieri dei partiti al governo (ad eccezione della consigliere FdI Anna Scarafoni, che sostiene il punto 3 della mia mozione), nonché i consiglieri dei partiti di opposizione TEAM K, Verdi e PD hanno votato contro la mia mozione.
A subire le conseguenze è la popolazione, che evidentemente non può fidarsi dei responsabili della sanità pubblica, se questi negano persino i dati del ns. Istituto pubblico di statistica ASTAT allo scopo di bocciare una mozione a loro scomoda.
La votazione in Consiglio si è svolta, come da me richiesto, separatamente e per appello nominale, poiché ogni consigliere/assessore deve assumersi, in modo trasparente nei confronti dei cittadini, la responsabilità della decisione contro la mia mozione.