HERBERT DORFMANN (Südtiroler Volkspartei) contribuisce – una volta di più – alla progressiva introduzione di meccanismi orwelliani nell’UE
Un’altra giornata nerissima al Parlamento europeo
Il Chat-Control (scansione in tempo reale prima della crittografia dei messaggi digitali degli utenti privati) è stato oggi approvato a forza nel Parlamento europeo tramite un espediente procedurale estremamente controverso, nonostante la maggioranza dei parlamentari europei avesse già votato due volte contro.
Herbert Dorfmann (Südtiroler Volkspartei) si è schierato – come al solito – anche oggi dalla parte di coloro che, senza battere ciglio, cancellano con nonchalance i Diritti e le Libertà fondamentali dei cittadini dell’UE.
Il risultato nominale della votazione odierna al Parlamento europeo (la maggioranza ha votato contro la deroga al diritto alla privacy per i messaggi privati digitali, ma a causa di un espediente procedurale – molto discutibile – sarebbe stata necessaria la maggioranza assoluta per arrivare alla sospensione del controllo delle chat… vedi in allegato i risultati della votazione per nome) ha conseguenze inaccettabili e pericolose, ed è un documento storico di fondamentale importanza, perché testimonia chi è a favore della creazione di un complesso di controllo e di censura del regime autoritario dell’UE, al quale Herbert Dorfmann collabora con grande zelo.
Le maggioranze democratiche non contano più al Parlamento europeo.
Si calpestano tutte le regole democratiche per privare – con delle motivazioni del tutto pretestuose – i cittadini sempre di più della loro privacy, libertà e il benessere.
Metà del Consiglio provinciale altoatesino-sudtirolese vota a favore dell’annullamento delle multe inflitte per il mancato uso della mascherina, multe palesemente illegittime per diversi motivi
Il governatore dell’Alto Adige – Sudtirolo e il governo provinciale altoatesino-sudtirolese (incluso Marco Galateo di FdI) dimostrano ancora una volta un’enorme difetto di consapevolezza dei Diritti Fondamentali ed insistono nell’incasso delle multe
Tutta l’opposizione presente oggi in Consiglio provinciale altoatesino (Team K, Südtiroler Freiheit, Verdi, Noi Cittadini, JWA, Freie Liste, Thomas Widmann) e la consigliera di Fratelli di Italia Anna Scarafoni hanno votato a favore della mia mozione 436/26, con la quale ho chiesto la sospensione immediata della riscossione delle sanzioni – inflitte in modo palesemente illegittimo dalla Segreteria Generale della Provincia Autonoma di Bolzano per il mancato uso della mascherina all’aperto- nonché l’annullamento di tutte le multe comminate e il rimborso di quelle già pagate:
Esito della votazione: 17 voti a favore della mozione, 17 voti contrari (vedi l’esito della votazione nominale in allegato).
Hanno votato contro: Marco Galateo (Fratelli d’Italia), tutti i consiglieri della Südtiroler Volkspartei, Christian Bianchi di Forza Italia, Angelo Gennaccaro della Lista Civica e Ulli Mair dei Freiheitlichen.
Le sanzioni pecuniarie sono palesemente illegittime per diversi motivi:
Il difetto di ragionevolezza e la nocività dell’obbligo di indossare la mascherinaall’aperto sono state nel frattempo espressamente confermate sia dall’Assessore alla Salute dell’Alto Adige, sia dal Governatore dell’Alto Adige – Sudtirolo nel corso della loro audizione in Commissione d’inchiesta del Consiglio provinciale dell’Alto Adige – Sudtirolo sulle misure anti-Covid. Ciò era del resto già noto nel 2020, come conferma la Camera dei Rappresentanti degli Stati Uniti nella sua relazione finale del 2024 sulle misure Covid:
La limitazione dei diritti fondamentali richiede sempre la verifica della sussistenza della sua necessità, utilità (ragionevolezza) e proporzionalità. L’imposizione dell’uso della mascherina per la prevenzione della circolazione del virus, in generale ma in particolare all’aperto, non è mai stata ragionevole e proporzionata.
Il governatore, nella sua dichiarazione odierna, ha paragonato al divieto di sosta con un autoveicolo la limitazione del diritto fondamentale di poter respirare liberamente all’aria aperta senza dover indossare la mascherina, dimostrando ancora una volta – sia come giurista che come governatore – un grave difetto nella consapevolezza della portata dei Diritti Fondamentali.
Il Governatore Kompatscher sostiene che non sarebbe necessario né opportuno annullare le sanzioni e sospendere la riscossione delle multe non ancora incassate, seppur la limitazione del diritto fondamentale si fosse rivelata essere del tutto irragionevole.
Arno Kompatscher – il presidente della Provincia Autonoma di Bolzano – con il sostegno degli altri membri del governo (tra i quali l’Assessore Marco Galateo di Fratelli d’Italia) continua a scagliare la Alto Adige – Sudtirolo Riscossione S.p.A. contro i cittadini che, legittimamente – anche per legittima difesa ai sensi dell’art. 4 della legge n. 689/1981 contro una misura disumana e irragionevole – si muovevano all’aperto senza mascherina.
La Segreteria generale della Provincia Autonoma di Bolzano non aveva il potere di infliggere tali sanzioni, poiché la Provincia Autonoma di Bolzano, nell’ambito della profilassi internazionale, poteva agire in modo autonomo solo se lo Stato non fosse già intervenuto con misure e, inoltre, solo se la situazione epidemiologica lo richiedesse (art. 2 del decreto legge n. 19/2020).
Per quanto riguarda l’uso della mascherina all’aperto, tuttavia, lo Stato centrale aveva già disposto misure in materia e, secondo le disposizioni statali (D.L. 19/2020 – artt. 3 e 4), il potere per l’irrogazione delle sanzioni pecuniarie spettava esclusivamente al prefetto e a nessun altro organo.
La Corte costituzionale, con sentenza n. 97/2025, ha stabilito che le sanzioni comminate dalla Segreteria generale della Provincia autonoma di Bolzano per il mancato uso della mascherina all’aperto devono essere valutate esclusivamente sulla base delle disposizioni nazionali (D.L. 19/2020 – art. 4).
Ciò significa che le sanzioni inflitte ai cittadini dalla Segreteria generale della Provincia Autonoma di Bolzano sono persino radicalmente nulle, poiché il legislatore nazionale non ha conferito alla Segreteria generale della Provincia Autonoma di Bolzano alcun potere generale per l’irrogazione delle sanzioni.
Anna Scarafoni (FdI), nel suo intervento sulla mia mozione, ha sottolineato che l’impegno assunto nel programma di coalizione del Governo provinciale altoatesino-sudtirolese di sottoporre a una revisione critica le misure anti-Covid, obbliga il Governo provinciale – alla luce dei fatti – ad annullare tali sanzioni pecuniarie. Ha quindi votato a favore della mia mozione, mentre il suo collega di partito di Fratelli di Italia, l’Assessore Marco Galateo – come al solito – non l’ha fatto, dimostrando ancora una volta cosa significhi l’incoerenza nella politica.
Così facendo, l’assessore Marco Galateo di Fratelli d’Italia ha votato affinché la Alto Adige – Sudtirolo Riscossione S.p.A. possa riscuotere presso i cittadini – con esecuzione forzata – sanzioni inflitte per non essersi attenuti ad un obbligo, la cui irragionevolezza è stata espressamente riconosciuta anche dal Governatore e dall’Assessore alla Salute.
Ma, la coerenza, correttezza e legalità in politica è una rarità.
L’irragionevolezza dell’obbligo di indossare la mascherina all’aperto è stata da tempo confermata a livello internazionale e ora anche dal governatore dell’Alto Adige-Sudtirolo e dall’Assessore provinciale alla sanità.
Ciononostante, il Governo provinciale altoatesino-sudtirolese continua a riscuotere dai cittadini le sanzioni inflitte – peraltro senza averne il potere – dal Segretariato generale della Provincia autonoma di Bolzano per il mancato uso della mascherina all’aperto, minacciando l’esecuzione coattiva.
Lo Stato di diritto non è certo questo!
Con la mia mozione n. 436/2026 ho invitato il Consiglio provinciale dell’Alto Adige-Sudtirolo e il Governo della Provincia Autonoma di Bolzano a ripristinare lo Stato di diritto.
Oltre al fatto che tanti faccendieri vicini alla politica si sono arricchiti senza scrupoli con il commercio delle “museruole” completamente inutili ai fini del contenimento della diffusione virale, si insiste in un’evidente violazione del Diritto Fondamentale dei cittadini al rispetto della propria Dignità Umana e dell’incolumità psico-fisica
Conferma da parte dell’Assessore alla Salute e del Governatore della Provincia Autonoma di Bolzano del difetto di ragionevolezza e proporzionalità dell’obbligo di portare la mascherina all’aperto
In occasione dell’audizione dell’Assessore alla Salute Dr.med. Hubert Messner in Commissione d’inchiesta sulle misure Covid del Consiglio della Provincia Autonoma di Bolzano, alla seguente domanda (tradotta dal tedesco in italiano):
“La prossima domanda riguarda il futuro piano pandemico. Argomento: mascherine. Sicuramente Lei conosce, tra gli altri, il metastudio Cochrane del 2023, che conferma in definitiva che non è stata riscontrata alcuna differenza nella trasmissione del virus con o senza mascherina. Pertanto, la mia domanda è: in caso di una prossima infezione virale – che speriamo non si verifichi – che dovesse assumere tali proporzioni, l’approvvigionamento di mascherine sarà una questione di principio e uno strumento fondamentale, nonostante non sia scientificamente provato che abbiano un effetto sulla trasmissione del virus?“
L’Assessore ha risposto testualmente come segue (tradotto dal tedesco in italiano):
“Per quanto riguarda le mascherine, è vero… Sappiamo che le mascherine non sono certamente la soluzione ideale. Non le useremo all’aperto, credo che questo non debba più succedere, non all’apertoma in spazi affollati.”
Vedi l’estratto del verbale della Commissione d’inchiesta d.d. 21.01.2026:
Il fatto che il governo provinciale nel nuovo piano pandemico non preveda più l’uso delle mascherine all’aperto è l’inequivocabile prova del fatto che l’imposizione dell’uso delle mascherine all’aperto era ed è sfornita di evidenza scientifica dell‘utilità e, dunque, della ragionevolezza e proporzionalità della misura imposta.
Anche il Governatore della Provincia Autonoma di Bolzano nell’audizione in Commissione d’inchiesta sulle misure Covid del Consiglio della Provincia Autonoma di Bolzano, che ebbe luogo il 4 giugno 2026, ha confermato il difetto di ragionevolezza e proporzionalità dell’obbligo di usare la mascherina all’aperto.
Il Governatore ha dichiarato in audizione da parte della Commissione d’inchiesta sulle misure del Covid testualmente quanto segue (tradotto dal tedesco in italiano):
«Per quanto riguarda l’obbligo di indossare la mascherina all’aperto, condivido l’opinione del collega Messner. Oggi non si prederebbe più una misura del genere. Già all’epoca quella misura era tra le più controverse tra le varie adottate.“
Il difetto di utilità dell’uso della mascherina ai fini della prevenzione della trasmissione virale era già stata confermata con uno studio meta da parte della rinomata rete tra scienziati a livello internazionale Cochrane.
Il difetto di utilità dell’uso della mascherina (soprattutto all’aperto, ma non solo) era ben noto già nel 2020 (e ancora prima) come risulta anche dal rapporto finale del U.S. House of Representatives(Camera dei Rappresentanti degli Stati Uniti) d.d. 4 dicembre 2024.
Dal rapporto finale del Comitato di controllo sulle misure covid del House of Representatives degli USA risulta testualmente (tradotto dall’inglese in italiano):
„Con il progredire della pandemia, hanno cominciato ad apparire ulteriori pubblicazioni scientifiche sottoposte a revisione paritaria sul tema delle mascherine. Nel maggio 2020, uno studio pubblicato su Emerging Infectious Diseases ha rilevato che “nell’analisi aggregata, non abbiamo riscontrato una riduzione significativa della trasmissione dell’influenza con l’uso delle mascherine“. In tale studio, i ricercatori hanno condotto una revisione della letteratura scientifica su diversi RCT relativi a diversi interventi non farmaceutici per gli studi sull’influenza pandemica, tra cui dieci sulle mascherine. Sempre nel maggio 2020, il New England Journal of Medicine ha pubblicato un articolo sull’uso delle mascherine negli ospedali: Queste ricerche hanno osservato che “sappiamo che indossare una mascherina al di fuori delle strutture sanitarie offre una protezione minima, se non nulla, dall’infezione …
Durante una deposizione relativa alla causa intentata dai procuratori generali della Louisiana e del Missouri, che accusa l’amministrazione Biden di collusione per censurare i discorsi sul COVID-19 sui social media, l’avvocato del querelante ha chiesto al dottor Fauci su quali studi si fosse basato il CDC per giustificare l’obbligo delle mascherine. Hanno chiesto al dottor Fauci quanti studi fossero stati condotti e se alcuni di essi fossero basati sul placebo …
Sono stati condotti studi randomizzati in doppio cieco tra febbraio 2020 e aprile 2020. Il dottor Fauci ha risposto di non ricordarlo. È assolutamente essenziale che queste decisioni, che hanno avuto conseguenze nella vita reale, possano essere verificate a posteriori.
Il dottor Fauci ha ammesso che, a livello di popolazione, le mascherine non forniscono una copertura efficace,…
Alla fine di gennaio 2023, Cochrane ha pubblicato la revisione più rigorosa e completa della letteratura scientifica sulle mascherine durante la pandemia di COVID-19. Cochrane è considerata l’organizzazione più rispettata al mondo per la valutazione degli interventi sanitari, è nota per essere la migliore risorsa per la ricerca metodologica ed è riconosciuta come l’organizzazione con il più alto standard di assistenza sanitaria basata sull’evidenza.
La pubblicazione del gennaio 2023 ha rilevato che indossare qualsiasi tipo di copertura per il viso “probabilmente fa poca o nessuna differenza” nel ridurre la diffusione delle malattie respiratorie. Lo studio ha esaminato 15 trial che confrontavano i risultati dell’uso di mascherine chirurgiche rispetto all’assenza di mascherine e anche rispetto alle mascherine N95, in ambito ospedaliero e comunitario durante la pandemia. La conclusione è stata che il valore dell’uso delle mascherine era approssimativamente pari a zero. “Non c’è alcuna prova che facciano alcuna differenza. Punto.
Le traiettorie del tasso di contagi da COVID-19 negli Stati che hanno imposto l’obbligo di indossare la mascherina e in quelli che non l’hanno fatto sono praticamente identiche. Undici Stati non hanno mai imposto l’obbligo di indossare la mascherina, mentre gli altri hanno adottato misure di applicazione in qualche forma …
È evidente che il CDC e l’amministrazione Biden hanno selezionato con cura i dati osservazionali per adattarli alla loro narrativa secondo cui le mascherine sono pienamente efficaci. Tuttavia, questo non è il ruolo del CDC. Il CDC è un’agenzia che ha lo scopo di proteggere il popolo americano e parte di tale responsabilità include la conduzione, la sponsorizzazione o, per lo meno, l’esame di studi clinici per disporre effettivamente delle migliori ricerche disponibili prima di formulare le proprie linee guida.“
Il difetto della ragionevolezza e proporzionalità dell’imposizione dell’uso della mascherina – a maggior ragione all’aperto, è, dunque, evidente e confermata a livello istituzionale.
Non solo l’inutilità, ma pure la nocivitàper la salute dei cittadini dell’imposto uso della mascherina è stata esposta con professionalità dal grande patologo Prof.Dr.med. Arna Burkhardt a marzo 2023.
Qui la versione oridignale del suo autorevole in lingua tedesca:
Il ricercatore di aerosol Prof. Gerhard Scheuch, noto a livello internazionale, già consulente del Robert Koch Institut durante la cosiddetta pandemia, nel 2023 ha confermato che l’uso delle mascherine non ha alcun impatto sulla diffusione virale:
Dalla confermata non lesività del comportamento dei cittadini sanzionati, e dalla nocività della misura con il rispettivo diritto die cittadini alla legittima difesa, deriva l’illegittimità – per violazione dei principi generali della Legge 689/1981 – degli ordini-ingiunzione emessi peraltro in difetto di competenza (ultra vires) dalla Segreteria generale della Provincia Autonoma di Bolzano
In base ai principi generali (legge n. 689/1981) elaborati dalla giurisprudenza, le sanzioni amministrative richiedono che la condotta sia non solo contraria alla legge (antigiuridica), ma anche concrettamente lesiva dell’interesse tutelato dalla norma.
L’interesse tutelato con l’imposto obbligo di usare la mascherina all’aperto sarebbe stato quello di contenere la diffusione del virus all’aperto.
A parte il fatto che le mascherine non hanno alcun impatto sulla la circolazione del virus (vedi sopra), tale necessità ed efficacia comunque non esiste all’aperto.
E poi, l’imposto uso della mscherina pure all’aperto ha avuto degli effetti negativi sulla salute psico-fisica die cittadini (vedi sopra) e, dunque, sussisteva anche il diritto alla legittima difesa ex art. 4 Legge n. 689/1981.
Dato che sia l’Assessore alla Salute, sia il Governatore della Provincia Autonoma di Bolzano hanno confermato in Commissione d’inchiesta sulle misure Covid il difetto di ragionevolezza e proporzionalità dell’imposto obbligo di usare le mascherine all’aperto, i cittadini che non hanno messo la mascherina all’aperto non hanno tenuto un comportamento concretamente lesivo dell’interesse che si ha inteso tutelare.
E, dunque, le rispettive ordinanze-ingiunzione erano e sono illegittime perchè con loro la Segreteria generale della Provincia Autonoma di Bolzano, peraltro senza averne la competenza, ha inflitto una sanzione ai cittadini per un comportamento che non era lesivo dell’interesso che si intendeva tutelare.
I Diritti Fondamentali sono assoluti e la loro limitazione richiede sempre la rigorosa prova della ragionevolezza e proporzionalità
L’imposizione ai cittadini dell’uso di una mascherina persino all’aperto – in difetto di comprovata utilità – costituiva una chiara violazione della dignità umana e dell’integrità psico-fisica.
Anche la violazione della dignità umana costituisce tortura.
L’imposizione – senza la previa dimostrazione della ragionevolezza e proporzionalità – dell’uso della mascherina, viola gli artt. 2, 13 e 32 Costituzione, l’art. 3 Cedu, nonché l’art. 4 della Carta dei Diritti e delle Libertà Fondamentali dell’UE.
Nullità delle sanzioni e delle rispettive ordinanze-ingiunzione inflitte/notificate ai cittadini per difetto assoluto di competenza in capo alla Segreteria della Provincia Autonoma di Bolzano
Con sentenza n. 97/2025 depositata il 3 Luglio 2025 la Corte Costituzionale ha dichiarato la questione di legittimità costituzionale sollevata dal Tribunale di Bolzano inammissibile, perché “L’ordinanza impugnata … nella parte motiva richiama esclusivamente l’art. 4 del d.l. n. 19 del 2020, come convertito, ossia la disposizione statale che sanzionava la violazione degli obblighi imposti durante la pandemia.
Ne consegue che … la sanzione contestata innanzi al giudice comune è stata irrogata facendo applicazione unicamente della legge statale.
Non dovendo il rimettente fare applicazioni delle disposizioni provinciali censurate, pertanto, le questioni di legittimità costituzionali sollevate sono inammissibili.”
Vedi qui la sentenza con la parte finale del testo decisivo evidenziato:
Poiché il Tribunale di Bolzano ha sollevato la questione di legittimità costituzionale esclusivamente in merito all’eccesso di competenza da parte del legislatore provinciale, la Corte costituzionale – visto il riferimento esclusivo, nella parte motiva della sanzione pecuniaria, all’art. 4 del D.L. 19/2020 – ha ritenuto che il Tribunale di Bolzano dovesse decidere la questione della legittimità dell‘ordinanza-ingiunzione applicando esclusivamente le disposizioni nazionali (D.L. 19/2020, artt. 1 e 4) – ha dichiarato l’inammissibilità (e non la infondatezza) della questione di legittimità costituzionale, poiché, secondo la Corte costituzionale, era superflua.
Il Tribunale provinciale di Bolzano non aveva sollevato la questione della legittimità costituzionale delle disposizioni nazionali (D.L. 19/2020, artt. 1 e 4) in relazione alla ragionevolezza e alla proporzionalità dell’obbligo di indossare la mascherina all’aperto, e pertanto la Corte costituzionale non si è pronunciata al riguardo.
La Segreteria generale della Provincia Autonoma di Bolzano aveva sì citato, nell’introduzione delle ordinanze-ingiunzione notifcate ai cittadini, anche la legge n. 4 della Provincia Autonoma di Bolzano dell’8 maggio 2020
e il provvedimento d’urgenza del presidente della Provincia rispettivamente in vigore, ma poi, nella parte motivativa vera e propria dell’ordinanza-ingiunzione, aveva fatto riferimento solo alla disposizione nazionale (decreto legge n. 19 del 25 marzo 2020). Ciò è stato determinante affinché la Corte costituzionale ritenesse che la normativa provinciale fosse irrilevante ai fini della decisione sul ricorso contro le ordinanze-ingiunzione emesse dal Segretario generale della Provincia autonoma di Bolzano.
Vista questa sentenza, bisogna verificare se la Provincia Autonoma di Bolzano – in persona del suo Segretario generale – sulla base della normativa nazionale era legittimata ad infliggere la sanzione prevista all’art. 4 del D.L. 19 del 25 marzo 2020 (sanzioni e controllo).
Ai sensi dell’art. 4 D.L. 25 marzo 2020 n. 19 (sanzioni e controlli ) “1. … il mancato rispetto delle misure di contenimento di cui all’articolo 1, comma 2, individuate e applicate con i provvedimenti adottati ai sensi dell’articolo 2, commi 1 e 2, ovvero dell’articolo 3, è punito con la sanzione amministrativa del pagamento di una somma da euro 400 a euro 1.000 ….
Le sanzioni per le violazioni delle misure di cui all’articolo 2, commi 1 e 2, sono irrogate dal Prefetto.
Stante la normativa statale (che tuttavia sono incostituzionali a causa della mancanza di ragionevolezza e proporzionalità dell’obbligo di indossare la mascherina all’aperto – in procedimenti ancora pendenti sono state presentate istanze di rinvio alla Corte costituzionale relative alla questione della costituzionalità, ma tali istanze non sono ancora giunte in questioni sollevate dal giudice e rinviate alla Corte costituzionale), il potere di irrogare la sanzione spetta solo al Prefetto e non alla Segreteria generale della Provincia Autonoma di Bolzano.
Dunque, gli atti di irrogazione della sanzione amministrativa emessi dalla Segreteria della Provincia Autonoma di Bolzano sono nulli per difetto assoluto di competenza.
Se il legislatore statale prevede espressamente un determinato organo per l’irrogazione della sanzione, un altro organo, che nella normativa nazionale (quella normativa che, secondo la Corte costituzionale, costituisce l’unica ed esclusiva base giuridica per le sanzioni inflitte con le ordinanze-ingiunzione emanate dalla Segreteria generale della Provincia Autonoma di Bolzano) non è previsto per l’irrogazione delle sanzioni, non può arrogarsi il potere di infliggere le sanzioni.
In questo caso si è evidentemente verificato un operato ultra vires (ovvero che esula dai poteri) da parte del Segretaria generale della Provincia Autonoma di Bolzano.
Gli ordini-ingiunzione notificati ai cittadini dalla Segreteria generale della Provincia Autonoma di Bolzano, con esclusivo riferimento nella parte motiva alla normativa statale, sono nulli per difetto assoluto di competenza.
Evidente difetto di legittimità costituzionale
degli artt. 1 e 4 D.L. 19/2020 in punto obbligo dell’uso della mascherina all’aperto
Anche per quanto da ultimo confermato dall’Assessore alla Salute e dal Governatore della Provincia Autonoma di Bolzano in sede istituzionale (vedi sopra), e per quanto risulta anche dal rapporto finale del Comitato sulla pandemia del coronavirus del U.S. House of Representatives, è evidente la violazione del principio di ragionevolezza e proporzionalità commessa con l’imposizione dell’ uso della mascherina all’aperto prevista nell’art. 1 D.L. 19/2020 e con la rispettiva sanzione nell’art. 4 D.L. 19/2020.
La restrizione dei Diritti Fondamentali, nel contesto costituzionale italiano ed europeo, non può essere lasciata all’arbitrio della politica, ma deve rigorosamente rispettare i principi di ragionevolezza e proporzionalità. Questi canoni costituiscono un limite al potere del legislatore e delle autorità, garantendo che le limitazioni siano necessarie, adeguate e proporzionate agli obiettivi pubblici perseguiti.
L’uso della mascherina all’aperto non soddisfa alcuno di questi criteri.
Nel caso dei diritti inviolabili (come è quello di girare liberamente senza doversi mettere una mascherina che blocca la naturale respirazione ed ha, dunque, effetti negativi di natura fisica, ma anche di natura psichica vista la copertura della parte emotiva del viso), le limitazioni sono possibili solo se bilanciate con altri interessi costituzionalmente protetti (es. dignità umana, salute, libertà), garantendo che il nucleo essenziale del diritto non venga compromesso.
In sintesi, una restrizione di un diritto fondamentale è legittima solo se è giustificata da un fine costituzionale, e se è adattaa conseguirlo, se è necessaria e proporzionata.
In Italia e nell’ordinamento europeo, affinché la limitazione di un diritto fondamentale sia legittima, non basta che sia prevista da una legge (riserva di legge), ma deve superare il cosiddetto test di proporzionalità.
I tre cardini di questo controllo sono:
Idoneità: la misura deve essere effettivamente capace di raggiungere l’obiettivo prefissato (un fine legittimo e meritevole).
L’uso di mascherine non ha alcun impatto significativo, persino nei locali chiusi come dimostra il meta-studio Cochrane.
Necessità: la restrizione deve essere la “misura minima” possibile. Se esiste un’alternativa meno invasiva che garantisce lo stesso risultato, va scelta quest’ultima. La situazione all’aria aperta rispetto a locali chiusi è radicalmente diversa riguardo alla circolazione e concentrazione virale. Dunque, la misura di imporre la mascherina all’aperto era evidentemente non necessaria.
Proporzionalità in senso stretto (Ragionevolezza): deve esserci un equilibrio costi-benefici. Il sacrificio richiesto al singolo non deve essere eccessivo rispetto al vantaggio ottenuto dalla collettività.
Dato che nel caso concreto, il vantaggio ottenuto dalla collettività non è solo zero, ma pure negativo (vedi impatto negativo sulla salute in generale per la collettività della misura), è ovvio che rimane solo il sacrificio richiesto al singolo cittadino.
In sintesi, il canone della ragionevolezzaevita che il potere legislativo agisca in modo arbitrario, trasformando la limitazione in una compressione intollerabile della dignità umana.
Nel caso concreto è evidente che l’imposto uso della mascherina all’aperto:
non era idoneo a raggiungere l’obbiettivo prefissato, che era quello di „contrastare e contenere la diffussione del virus“ (cfr. Epigrafe: … Ritenuta la strordinaria necessità e urgenza di emanare nuove disposizioni per contrastare l’emergenza epidemiologica da COVID-19, adottando adeguate e proporzoinate misure di contrasto e contenimento alla difussione del predetto virus“.
Per quanto riguarda la necessità,
A parte il fatto che la Segreteria della Provincia Autonoma di Bolzano ai fini dell’applicazione della sanzione nell’ordinanza impugnata „nella parte motiva richiama esclusivamente l’art. 4 del d.l. n. 19 del 2020“ (vedi sentenza della Corte Costituzionale n. 97/2025), ossia la disposizione statale che sanzionava la violazione degli obblighi imposti durante la pandemia, si evidenzia inoltre, che le disposizioni delle Ordinanze presidenziali contengibili e urgenti (che tuttavia non sono state invocate nella parte motiva die decreti-ingiunzione notificati ai cittadini dalla Segreteria Generale, come constatato dalla Corte costituzionale e che, secondo la stessa Corte, risultano pertanto irrilevanti) non corrispondono al presupposti di cui all’articolo 3 (misure urgenti a livello regionale o interregionale) del decreto legge n. 19 del 25 marzo 2020.
Infatti, la normativa nazionale (articolo 3 del decreto-legge n. 19 del 25 marzo 2020 – misure urgenti a livello regionale o interregionale) prevedeva che solo „nelle more dell’adozione die decreti del Presidente del Consiglio die ministri di cui all’articolo 2, comma 1, e con efficacia limitata fino a tale momento, le regioni, in relazione a specifiche situazioni sopravvenute di aggravamento del rischio sanitario verificatesi nel loro territorio o in una parte di esso, possono introdurre misure ulteriormente restrittive rispetto a quelle attualmente vigenti, tra quelle di cui all’articolo 1, comma 2, esclusivamente nell’ambito delle attività di loro competenza …“.
Il Presidente del Consiglio dei Ministri, ad esempio, aveva già disposto, con DPCM del 13 ottobre 2020 (DPCM), all’articolo 1, l’obbligo di indossare la mascherina:
Il fatto che in Alto Adige non ci fosse di un aggravamento, bensì di un miglioramento della cosiddetta situazione epidemiologica, comunicato al Governatore dall’Azienda sanitaria provinciale, e che egli abbia comunque disposto l’obbligo di indossare la mascherina all’aperto, risulta espressamente, ad esempio, dalle Ordinanze Presidenziali contingibili e urgenti n. 20 del 23/04/2021 e n. 25 del 18/06/2021.
Dalle premesse (CONSTATATO) emerge in entrambi i casi che la situazione epidemiologica è migliorata e non è peggiorata!
Così recita testualmente il provvedimento d’urgenza del governatore n. 20 del 23 aprile 2021: «CONSTATATO che in base a quanto riportato dal Direttore Generale e dal Direttore Sanitario dell’Azienda Sanitaria con nota dd. 23 aprile 2021, prot. N. 111058/21, considerata la diminuzione delle nuove infezioni sul territorio, il trend di riduzione della percentuale di positività ai tamponi e dell’indice Rt e della percentuale die posti letto occupati in area medica e nelle terapie intensive, la classificazione di rischio per la Provincia di Bolzano prefigura uno scenario di tipo 1° basso rischio di diffusione“.
Tuttavia, al punto 26, il Presidente della Provincia ha disposto quanto segue in merito alla “PROTEZIONE DELLE VIE RESPIRATORIE E DISTANZE DI SICUREZZA”:
«È fatto obbligo di avere sempre con se dispositivi di protezione delle vie respiratorie e di indossarli … in tutti i luoghi all’aperto, a eccezione die casi in cui, per le caratteristiche die luoghi o per le circostanze di fatto, sia garantita in modo continuativo la condizione di isolamento rispetto a persone non conviventi … ».
Nell’Ordinanza Presidenziale contingibile e urgente n. 25 del 18.06.2021 si legge testualmente: «CONSTATATO che, in base a quanto riportato dal Direttore Generale e dal Direttore Sanitario sostituto dell’Azienda Sanitaria con nota del 17.06.2021, prot. N. 168284/21, la situazione epidemiologica sul territorio della Provincia si è sviluppata in modo positivo …. In considerazione della situazione descritta, si ritiene possibile un allentamento delle misure di sicurezza in essere….“
Tuttavia, al punto 15 dell’Ordinanza (PROTEZIONE DELLE VIE RESPIRATORIE E DISTANZE DI SICUREZZA), il presidente della Provincia ha imposto testualmente quanto segue:
«È fatto obbligo di avere sempre con sé dispositivi di protezione delle vie respiratorie e di indossarli … in tutti i luoghi all’aperto, qualora non sia possibile mantenere la distanza interpersonale, e comunque in caso di assembramento…“
Alla luce dell’andamento epidemiologico comprovatamente positivo, non sussisteva certamente il presupposto indispensabile, previsto dall’art. 3 del decreto legge 19/2020, di una «situazione specifica di aggravamento del rischio sanitario, verificatasi nel territorio della provincia».
E poiché il legislatore statale aveva già adottato misure in merito tramite diversi decreti del Presidente del Consiglio dei Ministri (DPCM) (vedi sopra), il presidente della Provincia ha chiaramente oltrepassato le proprie competenze/poteri.
È evidente che le ordinanze-ingiunzioni notificate ai cittadini altoatesini-sudtirolesi sono nulle, poiché sono state emesse dalla Segreteria generale della Provincia autonoma di Bolzano in totale assenza del necessario potere a tal fine.
Dato che il vantaggio ottenuto dalla collettività non è soltanto ZERO, ma far girare la popolazione con le mascherine ha certamente avuto un impatto fisico e psichico (tutta la parte emotiva del viso è stata coperata) negativo, il risultato del test sulla proporzionalità è senz’altro negativo.
Le sanzioni inflitte ai cittadini altoatesini-sudtirolesi per i motivi sopra indicati violano gli artt. 2 (garanzia dei diritti inviolabili dell’uomo), 13 (inviolabilità della libertà personale) e 32 (invalicabilità del rispetto della persona umana) della Costituzione, degli artt. 1 (obbligo di rispettare i diritti dell’uomo), 3 (proibizione della tortura) della Convenzione Europea dei Diritti dell’Uomo nonché dell’art. 4 (proibizione di tortura) della Carta dei Diritti e della Libertà Fondamentali dell’UE.
Le disposizioni in parola, infatti, imponevano l’uso di un dispositivo di protezione delle vie respiratorie (mascherina) anche all’aperto, nonostante il radicale difetto della prova dell’utilità della misura imposta, e, dunque, in difetto di ragionevolezza e proporzionalità, di cui però deve essere caratterizzata ogni misura che comprime i Diritti e le Libertà Fondamentali.
Nonostante il difetto assoluto di competenza e nonostante l’ormai istituzionalmente dichiarato difetto di ragionevolezza e proporzionalità, la Segreteria della Provincia Autonoma di Bolzano insiste nell’esecuzione forzata tramite la Riscossione Alto Adige S.p.A. delle sanzioni inflitte ai cittadini per non aver usato la mascherina all’aperto.
Alla luce di quanto sopra esposto, si chiede alla Giunta della Provincia Autonoma di Bolzano nella persona del Governatore:
di voler subito comunicare all’Alto Adige Riscossione S.p.A. la sospensione dell’incarico per l’incasso delle sanzioni inflitte dalla Segreteria generale della Provincia Autonoma di Bolzano ai cittadini per non aver usato la mascherina all’aperto
di voler con urgenza ordinare alla Segreteria generale della Provincia Autonoma di Bolzano l’annullamento di tutte le sanzioni inflitte ai cittadini altoatesini-sudtirolesi per il mancato uso della mascherina all’aperto, e di voler, dunque, restituire ai cittadini altoatesini-sudtirolesi gli importi per le sanzioni da questi già pagati
2.1. perchè si tratta di sanzioni inflitte ultra vires in difetto assoluto di competenza della Segreteria generale della Provincia Autonoma di Bolzano per l’applicazione delle sanzioni amministrative di cui all’art. 4 D.L. 19/2020
2.2. perchè, in ogni caso, si tratta di sanzioni irrogate ai cittadini per un loro comportamento non lesivo e non offensivo dell’interesse tutelato (contrasto e contenimento alla diffusione del virus) e, dunque, di sanzioni irrogate in evidente difetto di ragionevolezza e proporzionalità.
Se per il periodo di allora il Governatore e i membri del Consiglio Provinciale che hanno votato a favore dell’applicazione da parte della Provincia Autonome di Bolzano di una sanzione ai cittadini per non aver usato la mascherina all’aperto, possono giustificarsi con l’ignoranza in punto difetto di ragionevolezza e proporzionalità della misura deliberata, oggi ogni membro di questo Consiglio Provinciale è – documenti alla mano – informato.
Il rigetto di questa mozione null’altro significherebbe che assumersi addesso – nonostante la piena consapevolezza – nell’attuale incarico di membro del Consiglio Provinciale e/o membro della Giunta Provinciale la responsabilità politica e giuridica di una espressa conferma di una misura autoritaria irragionevole, perchè non utile e dannosa.
La maggioranza del Consiglio della Provincia Autonoma di Bolzano (incluso Fratelli d’Italia) vuole vaccinare anche laddove l’OMS non lo ritiene necessario
In Alto Adige-Sudtirolo non vengono riconosciuti gli standard internazionali per la misurazione dello stato immunitario contro il tetano
La maggioranza del Consiglio provinciale altoatesino (incluso Fratelli d’Italia) vuole che i lavoratori e gli studenti vengano sospesi dal lavoro e dalle attività pratiche, anche se secondo gli standard internazionali sono considerati sufficientemente protetti contro il tetano
Più assurdo di così non si può!
Dal 1963 vige l’obbligo di vaccinazione antitetanica, tra l’altro, per i membri di determinate categorie professionali e per gli studenti delle scuole professionali e superiori che frequentano le lezioni pratiche.
Il tetano non è contagioso, non provoca pandemie, ma colpisce il singolo individuo. La vaccinazione protegge solo la persona vaccinata.
Grazie all’enorme miglioramento dell’igiene e dell’assistenza medica in Italia e in Europa nel XX secolo, il numero di casi di tetano in Italia era già diminuito rapidamente molto prima dell’introduzione dell’obbligo vaccinale contro il tetano (1963).
Un pericolo deriva da ferite profonde non pulite a regola d’arte. Di norma, il medico somministra agli feriti immunoglobuline e il vaccino antitetanico, indipendentemente dal fatto che siano già stati vaccinati in precedenza.
Secondo recenti notizie, in Italia il vaccino monovalente contro il tetano, che già ora è spesso difficile da reperire, non sarà più disponibile nel corso del 2026:
Già ora in Alto Adige-Sudtirolo molti adulti e adolescenti, che sono obbligati solo alla vaccinazione antitetanica, vengono vaccinati con un vaccino trivalente (tetano, difterite e pertosse) o con un altro vaccino multivalente.
Ciò non è consentito, poiché l’obbligo vaccinale (anche su questo argomento ci sarebbe molto da dire, ma non era oggetto della mozione) si riferisce esclusivamente al tetano.
Se i cittadini sono obbligati a vaccinarsi contro il tetano, non si può costringerli, per adempiere a tale obbligo, a farsi somministrare un vaccino combinato (multivalente) che non sia destinato a immunizzarli solo contro il tetano.
L’immunizzazione contro il tetano con un vaccino combinato, senza informare la persona interessata al riguardo, a meno che non sia lei stessa a sollevare specificatamente l’argomento, purtroppo è molto frequente.
È necessario garantire che le persone che sono dimostrabilmente protette dagli anticorpi antitetanici siano esentate dall’obbligo vaccinale.
Per determinare gli anticorpi è necessario il cosiddetto titolo antitetanico.
Il titolo antitetanico indica quanti anticorpi protettivi (IgG) contro la tossina tetanica sono presenti nel sangue.
Viene determinato tramite prelievo di sangue (siero).
La misurazione in laboratorio degli anticorpi IgG antitetanici avviene di norma mediante la procedura standard ELISA (Enzyme-Linked Immunosorbent Assay).
I risultati sono espressi in unità internazionali per millilitro (IU/ml o IE/ml).
Secondo le informazioni disponibili, il Laboratorio Centrale di Bolzano dell’Azienda Sanitaria Provinciale dell’Alto Adige utilizza il test ELISA EUROIMMUN del produttore Medizinische Labordiagnostika AG.
Secondo la descrizione del prodotto, con un valore di Ul/ml ≥ 0,1 è presente una protezione immunitaria che NON viene considerata insufficiente.
Il produttore del test ELISA utilizzato dall’Azienda Sanitaria dell’Alto Adige dichiara che una vaccinazione di richiamo garantisce una protezione vaccinale a lungo termine, ma NON RACCOMANDAla immediata vaccinazione di richiamo.
La vaccinazione di richiamo è raccomandata solo in caso di un valore di anticorpi inferiore a 0,1 UI/ml.
Pertanto, con un titolo anticorpale di ≥ 0,1 UI/ml non è raccomandata la vaccinazione di richiamo, ma si fa semplicemente notare che essa garantisce una protezione vaccinale a lungo termine.
Dalla tabella di interpretazione dell’Azienda Sanitaria Provinciale dell’Alto Adige risulta invece che, con un titolo compreso tra 0,11 e 0,5 UI/ml, è raccomandata la vaccinazione di richiamo.
Pertanto, la tabella di interpretazione dell’Azienda Sanitaria Provinciale dell’Alto Adige si discosta da quella del produttore del test ELISA utilizzato dall’Azienda Sanitaria Provinciale dell’Alto Adige.
E questo ha conseguenze significative per i cittadini interessati.
Infatti, questa raccomandazione, che non risulta dalle informazioni sul prodotto, fa sì che i lavoratori e gli studenti interessati con un titolo antitetanico compreso tra ≥ 0,1 e 0,5 IU/ml vengano classificati dai medici del servizio di medicina del lavoro come “non idonei” alle attività professionali o pratiche scolastiche da svolgere, e sono quindi costretti a vaccinarsi per poter svolgere il proprio lavoro o partecipare alle lezioni pratiche e adempiere all’obbligo scolastico, sebbene presentino una protezione immunitaria sufficiente contro il tetano, che non richiede un richiamo immediato, e che viene addirittura valutata buona da esperti riconosciuti, come l’ex direttore dell’Istituto di Profilassi e Medicina Tropicale dell’Università di Medicina di Vienna.
Ciò che l’Azienda Sanitaria dell’Alto Adige definisce “protezione di breve durata” (che secondo il produttore Serion dura in ogni caso due anni e che secondo la letteratura scientifica viene addirittura definita “buona protezione immunitaria”) non può portare all’esclusione degli studenti dalle lezioni pratiche e alla sospensione dei lavoratori dal posto di lavoro.
Tanto più che in caso di ferite, i medici somministrano comunque a titolo profilattico, indipendentemente da una precedente vaccinazione antitetanica, immunoglobuline e la vaccinazione antitetanica.
La vaccinazione in presenza di un titolo di antitossina superiore a 0,5 IU/ml può causare effetti collaterali.
Nella pratica quotidiana, tuttavia, il titolo viene raramente misurato di routine.
Per soddisfare l’obbligo vaccinale contro il tetano, viene invece richiesta una vaccinazione di richiamo ogni 10 anni.
E questa viene molto spesso effettuata con un vaccino combinato (in Alto Adige con un vaccino duale e trivalente) dall’Azienda Sanitaria Alto Adige, senza che vi sia una corrispondente prescrizione medica.
presentata al Consiglio provinciale dell’Alto Adige, ho invitato i membri del Consiglio provinciale dell’Alto Adige a voler deliberare quanto segue:
Alla luce di quanto sopra esposto, si chieda alla Giunta provinciale dell’Alto Adige, nella persona dell’Assessore alla Sanità, di voler garantire che:
i vaccini monovalenti contro il tetano siano disponibili in quantità sufficiente e che ai cittadini non venga somministrato un vaccino combinato senza averli informati e senza il loro esplicito consenso;
la mancata disponibilità di un vaccino monovalente contro il tetano sia confermata per iscritto dall’Azienda Sanitaria dell’Alto Adige alla persona interessata dall’obbligo vaccinale contro il tetano, qualora questa ne faccia richiesta;
un titolo anticorpale contro il tetano pari o superiore a 0,11 UI/ml sia confermato dall’Azienda Sanitaria dell’Alto Adige come protezione vaccinale sufficiente;
che alle persone interessate, quando viene loro richiesto di dimostrare di essere state vaccinate contro il tetano, venga espressamente segnalato che l’obbligo vaccinale decade qualora venga dimostrato in laboratorio un livello sufficiente di anticorpi IgG contro la tossina tetanica;
le persone interessate dall’obbligo di vaccinazione antitetanica siano informate che la determinazione del titolo anticorpale è possibile e opportuna per evitare vaccinazioni inutili e potenziali effetti collaterali;
per la vaccinazione sia comunque presente una prescrizione medica del vaccino specifico da somministrare alla persona interessata, come prescritto dall’art. 88 del D.Lgs. 219/2006.
La mia mozione è stata respinta con 16 voti a favore e 19 contrari.
Hanno votato contro l’applicazione di standard scientifici riconosciuti a livello internazionale i due rappresentanti di Fratelli d’Italia (Marco Galateo e Anna Scarafoni), i rappresentanti di Forza Italia, della Lista Civica, della Südtiroler Volkspartei e dei Freiheitlichen.
Ringrazio i rappresentanti del Team K, della Südtiroler Freiheit, dei Verdi, della Freie Liste, di JWA, di Noi Cittadini e del PD per aver approvato la mozione.
Si veda la votazione per appello nominale in allegato.
Si tenga presente che mentre i due rappresentanti di FRATELLI D’ITALIA hanno votato contro la mozione, il rappresentante del PD ha votato a favore!
Consiglio ai cittadini di insistere per ottenere il vaccino monovalente (vaccino antitetanico monovalente) e, in caso di indisponibilità del vaccino monovalente presso l’Azienda Sanitaria Provinciale, di richiedere all’Azienda Sanitaria una conferma scritta di tale inaccettabile circostanza.
Ciò è particolarmente importante per i cittadini soggetti all’obbligo di vaccinazione antitetanica. Se l’Azienda Sanitaria non offre vaccini monovalenti, non è possibile esigere l’adempimento dell’obbligo vaccinale.
Per le persone soggette all’obbligo di vaccinazione antitetanica, è inoltre consigliabile determinare il titolo anticorpale e, in caso di valore pari o superiore a 0,1 UI/ml, insistere con il medico del lavoro per ottenere l’esenzione dalla vaccinazione sulla base degli standard internazionali.
ll Consiglio provinciale altoatesino contribuisce consapevolmente a maggioranza all’occultamento degli eventi avversi dei cosiddetti «vaccini» anti-Covid-19: questo è imperdonabile!
Migliaia di altoatesini soffrono delle conseguenze del cosiddetto «vaccino» anti-Covid-19 e non sono pochi quelli che ne sono morti.
L’incidenza dei tumori è in aumento.
Ciononostante – o forse proprio per questo (?) – la politica provinciale (partiti di governo e anche parte dell’opposizione) non vuole che se ne riveli la causa, con ciò evitando di creare i presupposti per garantire una terapia adeguata alle persone colpite e impedendo che in futuro vengano applicate ai cittadini in campagne “vaccinali” altre sostanze sperimentali.
Questo è imperdonabile!
Nonostante che
l’audizione in commissione d’inchiesta sul Covid del Consiglio della Provincia Autonoma di Bolzano di esperti riconosciuti del sistema sanitario italiano (Prof.Dott.med. Mariano Bizzarri, Ordinario di Patologia Clinica, Medicina Spaziale e Medicina Sperimentale all’Università La Sapienza, Prof.Dott.med. Marco Cosentino, Ordinario di farmacologia medica presso l’Università dell’Insubria a Varese, e il dott. Maurizio Federico, direttore scientifico del Centro Nazionale per la Salute Globale presso l’Istituto Superiore di Sanità), nonché le risposte dell’Assessore alla Salute Hubert Messner a specifiche interrogazioni, abbiano evidenziato che anche in Italia/Alto Adige-Sudtirolo non esiste alcuna farmacovigilanza sui cosiddetti “vaccini” contro il Covid-19
l’audizione e i documenti istituzionali confermino che l’efficacia e la sicurezza dei cosiddetti “vaccini” Covid-19 non sono mai state dimostrate
le autorità sanitarie nascondino palesemente la mancanza dell’effetto desiderato e l’alto rischio per la vita e salute collegato a queste sostanze sperimentali (vedi la corrispondenza e-mail trapelata dell’AIFA, l’insabbiamento da parte del FDA reso pubblico in questi giorni negli Stati Uniti, ecc.),
ulteriori sostanze basate su RNA modificata vengano in continuazione autorizzate dall’EMA come “vaccini” (vedi il recente “vaccino” combinato contro l’influenza e il Covid-19 mCOMBRIAX di Moderna)
alle donne incinte (nonostante il drastico calo del tasso di natalità dal 2021), agli studenti di medicina ecc. i cosiddetti “vaccini” anti-Covid-19 continuino ad essere raccomandati,
oggi i consiglieri della Südtiroler Volkspartei, di Fratelli d’Italia (Marco Galateo), dei Freiheitlichen, di TEAM K (in riferimento al punto 1 della mozione), Forza Italia e Lista Civica hanno rigettato, con delle motivazioni assurde, la mia mozione https://api-idap.landtag-bz.org/doc/IDAP_796472.pdfcon la quale richiedevo l’istituzione urgente in Alto Adige-Sudtirolo di una farmacovigilanza attiva per i cosiddetti “vaccini” anti-Covid-19 e i cosiddetti vaccini a RNA modificata in generale, nonché la presentazione (nell’ambito della Conferenza permanente per i rapporti tra Stato, regioni e province autonome) al governo nazionale della urgente richiesta di voler istituire finalmente questa farmacovigilanza attiva a livello nazionale!
Franz Ploner (TEAM K) ha affermato che la farmacovigilanza sarebbe sufficientemente svolta a livello centrale dall’EMA e dall’AIFA, che non sarebbe opportuno farla a livello altoatesino, sebbene nella mozione fosse stato dimostrato, sulla base di documenti istituzionali,
che l’EMA ha autorizzato queste sostanze sperimentali come “vaccini”, contrariamente alla loro effettiva composizione e modalità d’azione (prodotti di terapia genica), senza che ne fosse mai stata dimostrata l’efficacia e la sicurezza
che i responsabili dell’AIFA hanno palesemente taciuto intenzionalmente i gravi effetti collaterali nonché la mancanza dell’effetto desiderato (vedi e-mail trapelate), così come hanno fatto e continuano a fare l’EMA a livello UE e la FDA negli Stati Uniti (vedi la relazione della commissione d’inchiesta sull’argomento pubblicata al Senato degli USA il 29.04.2026)
che la farmacovigilanza passiva, specialmente per i nuovi farmaci, è assolutamente insufficiente (è dimostrato che viene segnalato solo dall’1 al massimo il 10 per cento degli eventi avversi)
che, a causa del diabolico algoritmo stabilito dall’OMS a favore dei produttori di vaccini per determinare gli effetti collaterali delle vaccinazioni, in presenza anche di una sola possibile causa alternativa, la vaccinazione viene automaticamente esclusa come causa, e quindi di fatto gli effetti collaterali vengono riconosciuti ufficialmente molto raramente
in Italia, in Germania ecc. per i cosiddetti pazienti Long-Covid sistematicamente non viene preso in considerazione se siano stati “vaccinati” contro il Covid-19 o meno; ciò vale anche per l’Alto Adige, come confermato dall’assessore alla salute Hubert Messner in occasione della sua audizione nella settimana di aprile del Consiglio provinciale
che dalle commissioni medico-legali delle aziende sanitarie o dell’INPS e dai tribunali italiani, sulla base delle perizie medico-legali dei CTU, è già stato accertato il nesso causale tra i cosiddetti “vaccini” Covid-19 e gravi danni neurologici, come ad esempio la mielite trasversa, e alle persone colpite è stato riconosciuto, ai sensi della legge 210/1992, un indennizzo per “danno da vaccino”, sebbene tali patologie non siano ancora state inserite dall’EMA – a causa della farmacovigilanza ostacolata – nel catalogo ufficiale degli effetti collaterali
l’Azienda Sanitaria dell’Alto Adige gestisce un centro provinciale per la farmacovigilanza (https://centrofarmacovigilanzabolzano.it/it/), sebbene sotto la direzione di un responsabile afflitto da gravi conflitti di interesse (Prof. Ugo Moretti), che aveva propagandata la “vaccinazione” anti-Covid-19 dei bambini! Non è quindi vero che la farmacovigilanza non sia e non possa essere gestita a livello locale, come sostiene il consigliere Franz Ploner, ma abbiamo bisogno di responsabili competenti e privi di conflitti di interesse!
Per tutti i cittadini altoatesini risulta da registrazione digitale nell’Azienda Sanitaria se sono stati “vaccinati” contro il Covid-19, quante volte, con quale prodotto “vaccinale”, da quale lotto di “vaccino” è stata prelevata l’iniezione, ecc. Tutte queste informazioni sono a disposizione dell’Azienda Sanitaria dell’Alto Adige. Una farmacovigilanza retrospettiva non sarebbe quindi un problema per l’Azienda Sanitaria Alto Adige! Ma la maggioranza del Consiglio provinciale altoatesino non la vuole.
È incredibile quanto siano resistenti ai fatti non solo i membri del governo provinciale, ma anche i membri dell’opposizione, i quali – soprattutto se hanno una formazione medica – avrebbero dovuto già da tempo sollecitare una farmacovigilanza attiva.
Purtroppo è esattamente il contrario.
Il presidente della Provincia ha nuovamente relegato la mia mozione nell’ambito del complottismo e, così facendo – considerato che la mia richiesta di una farmacovigilanza attiva è sostenuta, tra gli altri, anche dagli esperti italiani di comprovata competenza auditi dalla Commissione d’inchiesta sul Covid dell’Alto Adige (Prof. Mariano Bizzarri, Prof. Marco Cosentino e il dott. Maurizio Federico) – ha de facto offeso e denigrato anche questi esperti.
Niente di nuovo, poiché l’arroganza, anziché un confronto oggettivo, contraddistingue il governatore in questa questione di vitale importanza.
L’assessore provinciale Hubert Messner, in teoria responsabile della sanità, ha letto come al solito una risposta preconfezionata e ha dimostrato ancora una volta la sua assoluta incompetenza.
A favore della mia mozione relativa alla richiesta di istituire una farmacovigilanza attiva e autonoma in Alto Adige-Sudtirolo per i cosiddetti “vaccini” anti-Covid-19 e per i vaccini a RNA modificata in generale, hanno votato i colleghi di NOI CITTADINI, JWA e Südtiroler Freiheit, mentre i consiglierei dei Verdi (Brigitte Foppa ha lasciato l’aula poco prima della votazione), Freie Liste, del PD e Anna Scarafoni (FdI) si sono astenuti. Oltre a Brigitte Foppa, anche Thomas Widmann non ha partecipato alla votazione.
A favore della mia mozione per un appello urgente, nell’ambito della Conferenza permanente tra Stato, regioni e province autonome, al governo centrale affinché istituisca con urgenza questa farmacovigilanza attiva, hanno votato i colleghi di WIR BÜRGER, JWA, Südtiroler Freiheit, Freie Liste, mentre i deputati dei Verdi (Brigitte Foppa ha lasciato l’aula poco prima della votazione), TEAM K, PD e Anna Scarafoni (FdI) si sono astenuti. Oltre a Brigitte Foppa, anche Thomas Widmann non ha partecipato alla votazione.
Il fatto che la presidente della Commissione d’inchiesta sulle misure del Covid del Consiglio provinciale altoatesino, che dopo aver sentito i massimi esperti italiani si era detta sconvolta per la mancanza di farmacovigilanza (così si è potuto leggere sulla stampa), non abbia poi ritenuto necessario partecipare alla votazione su questa mozione importante per la salute della popolazione altoatesina, è a dir poco “strano”.
Si veda il risultato della votazione nominale in allegato.
Posso solo raccomandare a ogni cittadino di non farsi più iniettare nulla di ciò per cui la politica fa propaganda, perché la politica (… comprese parti importanti dell’opposizione)
lascia i cittadini prima all’oscuro dei possibili effetti collaterali,
e poi da soli con gli eventi avversi senza una terapia adeguata,
e non fa nulla – nonostante sia a conoscenza della drammatica situazione – per impedire che sostanze sperimentali pericolose vengano utilizzate nei programmi di vaccinazione, arrivando persino alle donne incinte (e quindi ai nascituri).
Farmacovigilanza – un presupposto fondamentale per la sicurezza dei medicinali – per i vaccini di fatto inesistente
È necessario intervenire in particolare sui cosiddetti “vaccini” contro il Covid-19 e, in generale, sui cosiddetti “vaccini” a base di RNA modificata
Il massimo responsabile della farmacovigilanza in Alto Adige e in Veneto – Prof. Ugo Moretti – si trova in un inaccettabile conflitto di interessi
L’Alto Adige/Sudtirolo deve intraprendere immediatamente una strada autonoma!
L’audizione presso la Commissione d’inchiesta sul Covid del Consiglio provinciale dell’Alto Adige / Sudtirolo di massimi esperti del sistema sanitario italiano (Prof. Mariano Bizzarri, Professore Ordinario di Patologia Clinica, Medicina Spaziale e Medicina Sperimentale, Università La Sapienza, Prof. Marco Cosentino, professore ordinario di farmacologia medica all’Università dell’Insubria a Varese, e il dott. Maurizio Federico, direttore scientifico del Centro Nazionale per la Salute Globale presso l’Istituto Superiore di Sanità), nonché le risposte dell’Assessore alla Salute Hubert Messner a una mia interrogazione, hanno confermato che non esiste alcuna farmacovigilanza sui cosiddetti “vaccini” contro il Covid-19.
Ad esempio,si omette sistematicamente – e quindi intenzionalmente – di distinguere, nei cosiddetti “pazienti” da Long Covid, se i loro disturbi siano dovuti a una naturale infezione virale o a una cosiddetta “vaccinazione” contro il Covid-19. Nel secondo caso, infatti, dovrebbero essere classificati e trattati come pazienti post-vaccinazione.
Secondo l’Assessore alla Salute Messner non ci sarebbero prove di un nesso causale tra la “vaccinazione” anti-Covid-19 e gravi danni neurologici (come la mielite, ALS ecc.) malattie autoimmuni e tumori: è vero esattamente il contrario!
In Italia, ad esempio, casi di mielite trasversa sono già stati riconosciuti ai sensi della legge 210/1992 (diritto all’indennizzo da danno vaccinale) dalle commissioni medico-legali delle aziende sanitarie (Alto Adige/Trentino) e dall’INPS (nel resto del territorio nazionale), nonché da sentenze giudiziarie, come conseguenza del cosiddetto “vaccino” anti-Covid-19.
Solo che questo viene intenzionalmente nascosto! Anche dal nostro governo provinciale.
(vedi i dettagli documentati nella mia mozione allegata o qui nella versione bilingue pubblicata sul sito del Consiglio della Provincia Autonoma di Bolzano: https://api-idap.landtag-bz.org/doc/IDAP_796472.pdf )
Ciò non sorprende, dato che il massimo responsabile della “farmacovigilanza” nella Provincia Autonoma di Bolzano è il Prof. Ugo Moretti (responsabile della farmacovigilanza anche della Regione Veneto):
Egli si trova in un inaccettabile conflitto di interessi nei confronti della popolazione.
Ha infatti personalmente promosso la “vaccinazione” anti-Covid-19 per i bambini e difficilmente avrà interesse a che gli effetti collaterali di queste sostanze sperimentali operanti di fatto come una “terapia genica” vengano accertati nella loro reale entità:
Ugo Moretti, considerato che gran parte della popolazione, tra cui molti bambini e adolescenti, è stata trattata con queste sostanze sperimentali su base di tecnologia genetica, non è accettabile come responsabile della farmacovigilanza!
Nella commissione d’inchiesta sul Covid della Provincia Autonoma di Bolzano è stato audito anche l’ex capo tossicologo di Pfizer in Europa, il dott. med. vet. Helmuth Sterz.
Egli ha illustrato quali test sarebbero stati necessari, vista la natura effettiva dei cosiddetti “vaccini” Covid-19 (sostanze sperimentali composti e funzionanti come i prodotti di terapia genica), e quali test necessari a garantire efficacia e sicurezza non sono stati però effettuati dai produttori dei cosiddetti “vaccini” Covid-19.
Ha spiegato, ad esempio, che i ratti utilizzati per la sperimentazione sugli animali sono assolutamente inadatti, poiché i ratti femmina, ad esempio, non hanno un ciclo mestruale.
Nel frattempo, i tassi di natalità in tutto il mondo stanno diminuendo rapidamente dall’introduzione della cosiddetta “vaccinazione” contro il Covid-19. Anche in Alto Adige-Sudtirolo, nel 2025 era molto al di sotto del valore del 2019.
La sua grande competenza in qualità di ex capo tossicologo di Pfizer in Europa, nonché la descrizione dell’inconfondibile sperimentazione umana lanciata a livello mondiale, è stata nel frattempo diffusa in Europa e nel mondo grazie alla sua interrogazione, registrata in video, da parte del Prof. Dr. Stefan Homburg nella Commissione d’inchiesta sul coronavirus del Bundestag tedesco, e ha portato all’attenzione urgentemente necessaria:
Il 29 aprile 2026, al Senato degli Stati Uniti è stata presentata la relazione della maggioranza della Commissione d’inchiesta permanente, dalla quale emerge, sulla base della corrispondenza interna della FDA (Food & Drug Administration) resa pubblica, che dopo l’introduzione del cosiddetto Covid-19 – sono stati immediatamente segnalati eventi avversi gravissimi, anche letali (anche i bambini sono stati colpiti) nel sistema statunitense di segnalazione degli effetti collaterali dei farmaci VEARS, ma queste segnalazioni e questi dati sono stati sistematicamente nascosti in modo intenzionale:
Inoltre, pochi giorni fa, il braccio destro di Antony Fauci è stato arrestato negli Stati Uniti per aver insabbiato l’origine del virus SARS-CoV-2 (un evidente prodotto della cosiddetta ricerca gain-of-function):
Lo stesso insabbiamento dei gravissimi eventi avversi sta avvenendo in Europa (l’autorità competente è l’EMA – Agenzia europea per i medicinali) e nella stragrande maggioranza degli Stati membri dell’Unione Europea, come l’Italia (l’autorità competente è l’AIFA – Agenzia Italiana del Farmaco) e la Germania (PEI – Paul Ehrlich Institut), con il forte sostegno, purtroppo, di una giurisprudenza che non segue più i principi costituzionali.
In Italia, la corrispondenza interna dell’AIFA trapelata – di cui si è parlato per mesi nel 2023 alla televisione nazionale nel programma „Fuori dal Coro“ (Mario Giordano) su Rete 4 (Mediaset) e sul quotidiano “LaVerità”– ha rivelato come i responsabili dell’AIFA e del Ministero della Salute abbiano intenzionalmente insabbiato in modo criminale per “non uccidere il vaccino” l’inefficacia e la pericolosità (compresi i decessi) dei cosiddetti “vaccini” Covid-19, evidenti subito dopo l’avvio della cosiddetta “campagna vaccinale”.
Poiché il governo Meloni, con il suo ministro della «Salute» Orazio Schillaci, porta avanti il sistema di assoluta opacità, tra l’altro sui cosiddetti «vaccini» anti-Covid-19, e la magistratura (con poche eccezioni) in Italia sostiene questa criminale opacità, i cittadini (compresi gli altoatesini / sudtirolesi) continuano a trovarsi di fronte alle conseguenze intollerabili di questa intenzionale e concertata omissione di una farmacovigilanza invece indispensabile per la sicurezza dei medicinali.
E ciò è assolutamente irresponsabile, considerato che nell’UE vengono autorizzati e utilizzati in Italia sempre più cosiddetti “vaccini” a base di RNA modificata (si veda la recente autorizzazione del “vaccino” combinato influenza/Covid-19 mCOMBRIAX di Moderna)!
Gli esperti scientifici auditi dalla Commissione d’inchiesta sulle misure del Covid del Consiglio della Provincia Autonoma di Bolzano hanno criticato la mancanza di farmacovigilanza in Italia per i cosiddetti “vaccini” Covid-19, nonché le gravi conseguenze che ne derivano.
Hanno chiarito in modo molto netto che questa situazione insostenibile non è cambiata nemmeno sotto il governo Meloni.
La risposta dell’Assessore alla Salute Hubert Messner alla mia interrogazione sulla farmacovigilanza relativa ai cosiddetti “vaccini” Covid-19 ha confermato la mancanza della farmacovigilanza necessaria per la sicurezza dei medicinali.
Qui la registrazione della interrogazione e la risposta dell’Assessore:
Il Prof. Marco Cosentino (Professore ordinario di Farmacologia Medica) ha dichiarato, nel corso della sua audizione in Commissione d’inchiesta parlamentare sulle misure del Covid che i cosiddetti “vaccini” contro il Covid-19
sono di fatto terapie geniche,
possono provocare la produzione illimitata, in termini di tempo e quantità, della proteina spike tossica e citotossica in ogni cellula del corpo della persona così “trattata”,
nonché una lunga serie di gravi effetti collaterali, in parte già confermati ufficialmente (come ad esempio la miocardite).
In questa situazione estremamente pericolosa per la salute della popolazione, è evidente l’urgente necessità di una farmacovigilanza attiva, anche retrospettiva!
L’urgenza è assoluta,
considerato il fatto che il Piano Nazionale di Vaccinazione vienedeterminato dai politici sulla base delle raccomandazioni di un’OMS controllata dai produttori di vaccini e dai cosiddetti filantropi (come la Fondazione Gates, che a sua volta investe massicciamente nel business dei vaccini) e dai loro tirapiedi:
il “vaccino” combinato a RNA modificata contro l’influenza e il Covid-19 (mCOMBRIAX del produttore Moderna) è stato approvato dalla Commissione Europea il 20 aprile 2026 senza prove di efficacia e sicurezza e sarà utilizzato in Italia/Alto Adige-Sudtirolo:
ulteriori cosiddetti “vaccini” a base di mRNA saranno presto approvati e, nel complesso, si sta promuovendo il passaggio a queste sostanze de facto di terapia genica nell’ambito dell’agenda di immunizzazione 2030 dell’OMS, tra l’altro perché i produttori hanno scelto un processo di produzione di massa molto economico – ovviamente con un alto rischio di residui di DNA pericolosi per la salute – e le autorità farmaceutiche lo lasciano passare in modo criminale
il tasso di natalità è in particolare forte calo dal 2021
non si può sperare nell’intervento della magistratura e delle autorità nazionali per evidenti ragioni intrinseche al sistema.
È giunto il momento che noi, in quanto responsabili politici per la salute dei cittadini altoatesini – sudtirolesi, facciamo ciò che in una situazione del genere è il minimo indispensabile: istituire immediatamente una farmacovigilanza autonoma e attiva!
E parallelamente dobbiamo esortare le autorità nazionali ad adempiere a questo loro dovere fondamentale nei confronti dei cittadini, atteso ormai da anni!
Per questo motivo ho presentato in Consiglio provinciale dell’Alto Adige/Sudtirolo la seguente mozione, che questa settimana sarà discussa in Consiglio e sottoposta a votazione:
Il Consiglio provinciale dell’Alto Adige/Sudtirolo incarichi la Giunta provinciale,
nella persona del Presidente della Provincia e dell’Assessore alla Salute, a istituire un sistema di farmacovigilanza attivo altoatesino/sudtirolese (anche retrospettivo), in aggiunta all’attuale sistema passivo e assolutamente insufficiente, specificamente per i cosiddetti “vaccini” Covid-19 e in generale per i cosiddetti “vaccini” a RNA modificata, sotto la guida di esperti non condizionati da conflitti di interesse
a impegnarsi, nella persona del Governatore, a richiedere con urgenza alla Presidente del governo di voler convocare, ai sensi dell’art. 12 della legge n. 400 del 23.08.1988, la Conferenza permanente per i rapporti tra Stato, Regioni e Province autonome, al fine di adottare con urgenza la necessaria delibera sull’ istituzione di un sistema attivo di farmacovigilanza (anche retrospettivo) relativo ai cosiddetti “vaccini” contro il Covid-19 e, in generale, ai cosiddetti “vaccini” a RNA modificata, sotto la guida di esperti che non siano in conflitto di interessi con il diritto fondamentale dei cittadini a medicinali efficaci e sicuri.
Per i tanti dettagli istituzionalmente documentati nelle premesse della mia mozione n. 407/26 in lingua italiana vedi l’allegato, e qui la versione bilingue pubblicata sul sito web del Consiglio provinciale dell’Alto Adige – Sudtirolo:
Anche i rappresentanti di FRATELLI D’ITALIA – come l’intera Giunta della Provincia autonoma di Bolzano (Südtiroler Volkspartei / Fratelli d’Italia / Forza Italia / Lista Civica e Freiheitliche) nonché Verdi e Für Südtirol con Widmann – non ravvisano alcun problema nel fatto che persino i Medici Direttori degli ospedali pubblici altoatesini dichiarano apertamente di omettere sistematicamente la lettura delle informazioni tecniche (Riassunto delle Caratteristiche del Prodotto medicinale – RCP) relative ai medicinali e ai dispositivi medici da loro utilizzati / prescritti / impiegati
La politica sanitaria altoatesina ha toccato il fondo
I rappresentanti di Fratelli d’Italia come l’intero Governo provinciale altoatesino (Südtiroler Volkspartei, Fratelli d’Italia, Forza Italia, Lista Civica e Freiheitliche) con il voto contrario alla mia mozione n. 390/26 hanno dimostrato di non ravvisare alcun problema nel fatto che persino i Medici Direttori degli ospedali pubblici dichiarino apertamente di omettere sistematicamente la lettura delle informazioni tecniche (Riassunto delle Caratteristiche del Prodotto medicinale – RCP) relative ai medicinali (farmaci e vaccini) e ai dispositivi medici da loro utilizzati / prescritti / applicati.
I Verdi e Thomas Widmann di Südtirol con Widmann si sono astenuti, omettendo così di esprimere il loro voto a tutela dei cittadini da errati trattamenti farmacologici e non farmacologici, programmati a questo punto, visto che i medici non si informano come invece dovrebbero per imposizione giuridica e medico-etica (Codice Deontologico art. 13) fare.
Tutti gli altri colleghi del Consiglio provinciale altoatesino/sudtirolese (i rappresentanti di TEAM K, Südtiroler Freiheit, Wir Bürger, JWA, Freie Fraktion e anche del PD) hanno votato a favore della mozione da me presentata, e dunque vorrei ringraziare i colleghi.
È assurdo che i due membri della Giunta provinciale altoatesina appartenenti al partito di Fratelli d’Italia (Marco Galateo e Anna Scarafoni) votino contro una mozione più che ragionevole, anzi – vista la situazione scandalosa che si è rivelata durante le audizioni in Commissione d’inchiesta sulle misure Covid – senz’altro a tutela dei cittadini dovuta!
Però, qui in Alto Adige / Sudtirolo vediamo in continuazione un comportamento di votazione in Consiglio provinciale dei rappresentanti di Fratelli d’Italia certo non consono alla tutela della salute dei cittadini.
Vedi la votazione per appello nominale sotto in allegato.
Le incredibili dichiarazioni rese nel corso dell’audizione in Commissione d’inchiesta sulle misure Covid del Consiglio della Provincia Autonoma di Bolzano dai medici direttori di tre ospedali altoatesini/sudtirolesi hanno portato alla luce uno scandalo enorme: persino i medici responsabili degli ospedali pubblici non si sentono in dovere di leggere le informazioni tecniche (riassunto delle caratteristiche del prodotto medicinale, RCP) sui medicinali e sui dispositivi medici (ad es. protesi) prima di utilizzarli sui pazienti o di raccomandarli su larga scala.
Che questa sia una situazione insostenibile, che mette a rischio la sicurezza dei pazienti / vaccinandi, dovrebbe essere chiaro a tutti, ma evidentemente non ai rappresentanti di Fratelli d’Italia, Forza Italia, Lista Civica, Südtiroler Volkspartei, Freiheitliche, Grüne e Südtirol mit Widmann.
La base giuridica dell’obbligo dei medici di informarsi adeguatamente su un medicinale o un dispositivo medico prima dell’uso / raccomandazione / prescrizione, nonché le conseguenze che comporta una violazione di questo chiaro dovere medico, sono state illustrate in modo documentato nella mia mozione n. 390/26 terminando nella seguente richiesta:
«Il Consiglio della Provincia Autonoma di Bolzano impegni la Giunta Provinciale, nella persona dell’Assessore alla Salute, a voler
ricordare – in forma adeguata (comunicato stampa, lettera aperta all’Ordine dei medici ecc.) – a tutti i medici che esercitano in Alto Adige/Sudtirolo che devono, nell’interesse della salute pubblica e dei loro pazienti, adempiere al loro obbligo di informarsi adeguatamente sulla natura, l’efficacia e la sicurezza di un medicinale prima di raccomandarlo, prescriverlo o somministrarlo
chiedere all’Azienda Sanitaria dell’Alto Adige, nella persona del Direttore Generale, che questa, nella sua qualità di datore di lavoro ossia committente, voglia ricordare per iscritto ai medici impiegati presso l’Azienda Sanitaria dell’Alto Adige il loro obbligo di informarsi adeguatamente sulla natura, l’efficacia e la sicurezza di un medicinale prima di raccomandarlo, prescriverlo o somministrarlo, nell’interesse della salute pubblica e dei propri pazienti.”
(vedi qui dalla pagina 28 in poi il testo in lingua italiana, oppure il documento solo in lingua italiana sotto in allegato):
Sebbene persino i medici direttori degli ospedali pubblici altoatesini dichiarano di sistematicamente omettere la lettura delle informazioni tecniche (Riassunto delle Caratteristiche del Prodotto medicinale – RCP), i rappresentanti di Fratelli d’Italia, Forza Italia, Lista Civica, Südtiroler Volkspartei, Freiheitliche, Verdi e Südtirol mit Widmann non ravvisano alcuna necessità di intervenire, esponendo così i cittadini al gravissimo rischio di errati trattamenti farmacologici e non farmacologici.
La politica sanitaria altoatesina ha toccato il fondo.
Se i direttori medici di ospedali pubblici consapevolmente non si informano sui medicinali e sui dispositivi medici prima di utilizzarli, la politica deve intervenire
Le incredibili dichiarazioni rese nel corso dell’audizione in Commissione d’inchiesta sulle misure Covid del Consiglio della Provincia Autonoma di Bolzano dai direttori medici di tre ospedali altoatesini/sudtirolesi hanno portato alla luce uno scandalo enorme: persino i medici responsabili degli ospedali pubblici non si sentono in dovere di leggere le informazioni tecniche (riassunto delle caratteristiche del prodotto medicinale, RCP) sui medicinali e sui dispositivi medici (ad es. protesi) prima di utilizzarli sui pazienti o di raccomandarli su larga scala.
Che questa sia una situazione insostenibile, che mette a rischio la sicurezza dei pazienti, dovrebbe essere chiaro a tutti.
La base giuridica dell’obbligo dei medici di informarsi adeguatamente su un medicinale o un dispositivo medico prima dell’uso / raccomandazione / prescrizione, nonché le conseguenze che comporta una violazione di questo chiaro dovere medico, sono illustrate in modo documentato nella mia mozione n. 390/26 con la richiesta rivolta al Governo della Provincia Autonoma di Bolzano di voler tra l’altro, provvedere affinchè l’Azienda Sanitaria dell’Alto Adige ricordi per iscritto ai medici ivi impiegati il loro dovere di informarsi adeguatamente sui medicinali prima di applicarli, considerando come livello minimo di informazione le informazioni tecniche ufficiali sul rispettivo prodotto medicinale (Riassunto delle caratteristiche del prodotto medicinale – RCP) fornite dall’Autorità del Farmaco al personale sanitario e pubblicate per dare accesso a tutti.
(vedi qui dalla pagina 28 in poi il testo in lingua italiana, oppure il documento solo italiano in allegato):
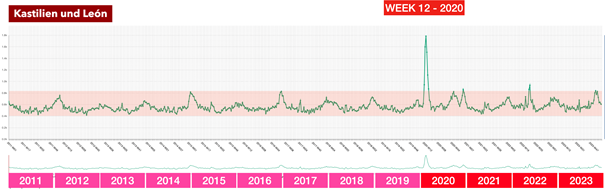
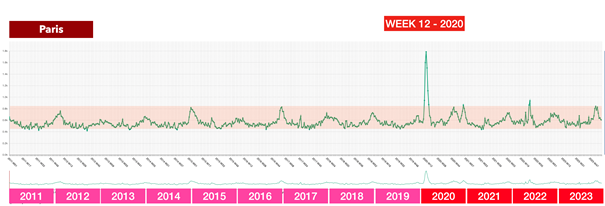
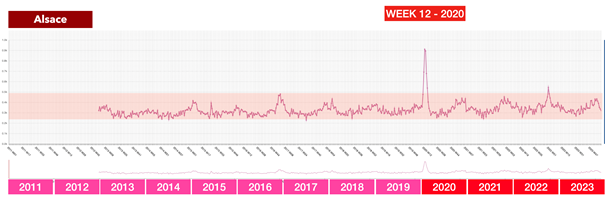
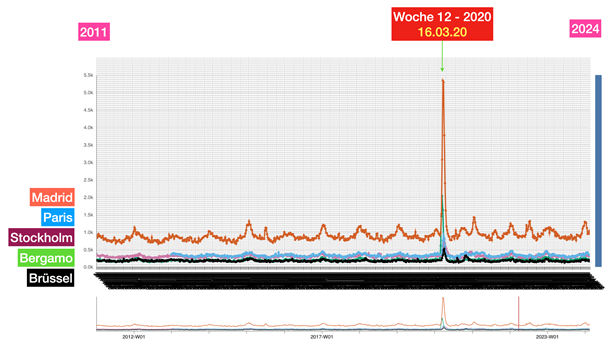
La riunione dei ministri della Salute tenutasi a Roma il 25 febbraio 2020 – l’evento chiave che ha portato successivamente all’adozione delle dannose misure anti-Covid in Italia e nella maggior parte dei paesi europei
Chi ha “guidato” i ministri della Salute?
A un esame più attento, l’incontro dei ministri europei della Salute tenutosi a Roma alla fine di febbraio 2020 appare non solo come una svolta, ma anche come un evento chiave descritto in modo altamente contraddittorio dal punto di vista politico.
Già sulla questione di chi abbia effettivamente dato il via a questo incontro, le versioni divergono: Roberto Speranza descrive nel suo libro che, dal suo punto di vista, l’incontro è nato in risposta all’aggravarsi della situazione nel Nord Italia, presentandosi così come un attore attivo che informa e coinvolge gli altri Stati europei.
A ciò si contrappone però la versione di Alain Berset, il quale afferma chiaramente che l’incontro è avvenuto su iniziativa di Jens Spahn, il quale avrebbe insistito per discutere la situazione a livello ministeriale, in particolare per quanto riguarda le misure alle frontiere e le reazioni politiche.
Non si tratta di un dettaglio, ma di una contraddizione fondamentale, poiché a seconda di quale versione sia corretta, ne emerge un quadro completamente diverso: o l’Italia si trovava in una situazione oggettivamente in escalation e ha invitato l’Europa a coordinarsi, oppure la Germania – sebbene a malapena colpita – ha attivamente promosso un’escalation politica a livello europeo. Entrambe le versioni non possono essere vere contemporaneamente.
Questa contraddizione diventa ancora più grave se si considerano i dati effettivi disponibili al momento di tale riunione: secondo l’analisi dei rischi dell’ECDC del 23 febbraio 2020, il rischio per la popolazione in Europa era solo «da basso a moderato», il numero di casi era limitato e non si prevedeva espressamente un sovraccarico dei sistemi sanitari.
In altre parole: l’autorità europea competente non vedeva alcuna catastrofe imminente. Eppure i partecipanti a quell’incontro riferiscono di aver “percepito fisicamente” che la situazione fosse fuori controllo. Qui si apre un enorme divario tra percezione e realtà – e questo divario non è stato colmato per caso.
Infatti, subito dopo questo incontro, in tutta Europa è iniziata una seconda ondata di influenza: modelli matematici che in quasi tutti i paesi lavoravano con drastici scenari da “worst case” e venivano presentati ai decisori politici come base per l’azione.
Il messaggio era strutturato allo stesso modo ovunque e presentava una notevole concordanza: se non si fosse proceduto immediatamente alla chiusura, in brevissimo tempo si sarebbero verificati numeri catastrofici di decessi. In Svezia erano previsti fino a 85.000 morti, nel Regno Unito fino a 500.000, in Germania circa 400.000 e in Svizzera circa 100.000. Queste cifre non sono state comunicate come ipotesi teoriche estreme, ma inserite nel processo decisionale politico come scenari realistici.
Ciò ha creato una pressione enorme sui ministri della sanità di tutta Europa.
Che si descriva questo come una «consulenza» o come un’effettiva delega da parte dei modellisti è, in definitiva, secondario: ciò che conta è che il processo decisionale politico si è disaccoppiato in brevissimo tempo dai dati reali per orientarsi invece verso scenari catastrofici ipotetici e modellizzati. Istituzioni come la Fondazione Bruno Kessler hanno fornito proprio questo tipo di modelli, che non servivano principalmente a descrivere la realtà, ma a giustificare e guidare le misure politiche.
Parallelamente, in Italia è iniziata un’applicazione sostenuta dallo Stato di farmaci non sufficientemente testati, tra cui combinazioni antivirali come Lopinavir/Ritonavir, che sono state ampiamente utilizzate nonostante la mancanza di evidenze – di fatto un esperimento medico su larga scala in condizioni reali.
Nel quadro d’insieme, il quadro si cristallizza in un’escalation strutturale: un incontro di origine politica, o almeno strumentalizzato politicamente, la cui paternità viene presentata in modo contraddittorio, si scontra con una valutazione del rischio oggettivamente moderata, è immediatamente seguito da modelli diffusi in modo sincronizzato in tutta Europa con previsioni di mortalità estreme ed è accompagnato da interventi medici sperimentali. Da questa combinazione nasce, nel giro di pochi giorni, una dinamica che coinvolge tutta l’Europa e spiana la strada a lockdown, chiusure scolastiche e profondi interventi nella vita sociale.
Il punto cruciale è questo: l’escalation non è stata innescata da dati chiaramente documentati, ma da un’interazione tra iniziativa politica, impressioni soggettive e scenari di worst-case modellizzati, che sono stati posti come base decisionale. Il fatto che ancora oggi non sia stato chiarito in modo univoco chi abbia effettivamente avviato questo incontro cruciale sottolinea quanto poco trasparente fosse la base di quelle decisioni che hanno poi riguardato milioni di persone.
Le conclusioni e i dati dell’esperto di analisi dati Tom Lausen, membro della commissione d’inchiesta del Bundestag tedesco (Enquete-Kommission des Deutschen Bundestages: https://www.bundestag.de/ausschuesse/weitere_gremien/ee01/1107006 -1107006) devono essere trasmessi a ogni commissione d’inchiesta europea sulle misure Covid, in primo luogo alla Commissione parlamentare di inchiesta sulla gestione dell’emergenza sanitaria da SARS-CoV-2 del Parlamento della Repubblica Italiana, poiché è in Italia che ha avuto origine lo strumentalizzato scenario apocalittico, le cui cause vanno accertate fino in fondo.
RA/Avv. DDr. Renate Holzeisen
Abgeordnete zum Südtiroler Landtag – Membro del Consiglio della Provincia Autonoma di Bolzano
Picchi di mortalità a Bergamo e in altre località dell’UE non spiegabili con una pandemia
Nell’ambito della sua audizione in Commissione d’inchiesta sulle misure Covid del Consiglio della Provincia Autonoma di Bolzano, l’analista di dati Tom Lausen ha dimostrato, sulla base di dati statistici ufficiali (Eurostat), che gli straordinaripicchi di mortalità registrati a Bergamo e in alcune altre località dell’Unione Europea non sono spiegabili con le conseguenze naturali di una pandemia.
I dati sulla mortalità a Bergamo e in numerose regioni europee nella primavera del 2020 mostrano un andamento che, in questa forma, non è né epidemiologicamente plausibile né è stato finora sufficientemente spiegato.
Per anni, la mortalità settimanale a Bergamo è rimasta stabile a circa 190 decessi. Nel marzo 2020 si è poi verificato un picco estremo e singolare, in cui i decessi sono aumentati di molte volte nel giro di pochi giorni – per poi tornare immediatamente al livello originario. Un andamento del genere è estremamente insolito per un evento infettivo. Le epidemie classiche mostrano ondate, non picchi verticali con normalizzazione immediata.
La domanda centrale è quindi: quale evento genera un picco così breve e allo stesso tempo massiccio, senza alcun proseguimento duraturo?
Per escludere che si tratti di errori statistici, l’analista di dati Tom Lausen ha condotto una ricerca sul posto a Bergamo insieme a un team di ricercatori. In 27 cimiteri sono state contate e documentate migliaia di tombe. Il risultato è inequivocabile: i dati sui decessi sono corretti. Non si tratta di un artefatto statistico. Il picco è reale. Ciò sposta radicalmente la questione: dalla qualità dei dati alle cause effettive di questo evento.
Particolarmente scottante è il fatto che Bergamo non sia un caso isolato. A Madrid, Parigi, in Alsazia e in Castiglia e León si osservano modelli simili quasi contemporaneamente nella settimana 12 del 2020: picchi di mortalità brevi ed estremi che non proseguono in ondate di lunga durata.
Questa sincronia tra diversi paesi richiede una spiegazione. Sistemi sanitari diversi, strutture demografiche diverse e misure politiche diverse non portano normalmente a picchi identici e puntuali, ma piuttosto a andamenti differenziati. È proprio questa differenziazione che qui manca.
Dopo il picco, i dati sulla mortalità in tutte le regioni considerate tornano rapidamente a un livello corrispondente alla media pluriennale. A Bergamo si registrano nuovamente circa 190 decessi a settimana.
Se la causa fosse stata esclusivamente un virus, ci si chiede perché, dopo un’epidemia così esplosiva, esso praticamente scompaia senza lasciare una fase prolungata di mortalità elevata. Un comportamento del genere è difficilmente conciliabile con i modelli epidemiologici conosciuti.
Ciò porta l’attenzione su altri fattori di influenza che finora non sono stati sufficientemente studiati. Tra questi figurano le strategie di trattamento medico nella fase iniziale, possibili errori terapeutici, carenze nell’assistenza, interruzioni delle cure mediche regolari e la gestione dei gruppi particolarmente vulnerabili, specialmente nelle strutture di assistenza.
Si pone inoltre la questione se le decisioni politiche e le misure organizzative possano aver contribuito esse stesse a questa dinamica.
Questi aspetti non sono questioni marginali, ma centrali per la comprensione degli eventi osservati.
I dati disponibili impongono una rivalutazione. Non si tratta più di stabilire se ci sia stato un picco di mortalità eccezionale – questo è indiscutibile e empiricamente provato. Si tratta di capire perché abbia assunto proprio questa forma: estremamente ripido, estremamente breve e senza un seguito duraturo. La spiegazione finora dominante, secondo cui un virus di nuova generazione avrebbe causato da solo questo andamento, è insufficiente e lascia aperte questioni essenziali.
Chi non pone queste domande rinuncia alla ricerca della verità. Chi le pone non può fare a meno di verificare anche ipotesi scomode. Questo è esattamente il compito di un’analisi scientifica e sociale.
I dati sottostanti provengono dalla banca dati ufficiale di Eurostat e sono accessibili al pubblico.
Settimana 12 – 2020
16.03.20
Sovramortalità in % (2020, settimana 1-25)
30 dicembre – 21 giugno 2020
Dato di riferimento = 2.587.569, scostamento = + 175,731 morti (+ 6,8 %)
Poiché il grafico copre il primo semestre del 2020, è chiaro che questi cluster di mortalità in eccesso si riferiscono a un periodo molto breve di poche settimane e non sono il risultato di un aumento persistente della mortalità nel corso di mesi.
Poiché in particolare l’eccezionale andamento della mortalità a Bergamo (che si è verificato solo con l’imposizione del lockdown, come dimostrato da Tom Lausen sulla base dei dati statistici ufficiali nella Commissione d’inchiesta sul coronavirus della Provincia Autonoma di Bolzano – cfr. comunicato stampa di ieri) è stato strumentalizzato come scenario da incubo per l’estensione a livello europeo di misure anti-Covid anche letali e comunque dannose a lungo termine per le persone, la società e l’economia, e i dati statistici e la loro evoluzione impongono le domande sollevate dall’analista di dati Tom Lausen, spetta a ogni Commissione d’Inchiesta sul Covid in Europa approfondire queste questioni/domande. E, in particolare, alla Commissione d’inchiesta del Parlamento Italiano!
Mi auguro, dunque, che la Commissione d’inchiesta vorrà invitare in Commissione d’inchiesta del Parlamento italiano l’esperto di analisi dati Tom Lausen (peraltro nominato a membro della Enquete-Kommission del Deutsche Bundestag (commissione d’inchiesta del parlamento germanico) sulle misure Covid fino al 2027 nel suo ruolo di esperto
Infatti, se non conosciamo le cause di questi eventi di mortalità straordinari e isolati, è impossibile raggiungere le conclusioni giuste per agire in futuro in modo ragionevole e adeguato nell’interesse dei cittadini.
RA/Avv. DDr. Renate Holzeisen
Abgeordnete zum Südtiroler Landtag – Membro del Consiglio della Provincia Autonoma di Bolzano